Chapter 1 Introduction
1.1 Denitrification and the nitrogen cycle
Many chemical elements of biological importance are cycled through the biosphere by a combination of physical and biological processes, in what are termed biogeochemical cycles. One of these cycles is the nitrogen cycle shown schematically in Figure 1.1.
![The nitrogen cycle. The oxidation state of nitrogen in each intermediate is given in brackets. Denitrification is shown by black arrows, nitrogen fixation by a dark grey arrow and all other reactions by light grey arrows. Note that nitrification involves two reactions, oxidation of ammonia to nitrite and of nitrite to nitrate, but these have never been found together in the same organism. ANAMOX (anaerobic ammonia oxidation) is a poorly-characterised process in which nitrite and ammonia combine to give water and dinitrogen [@fry_nitrogen_1992].](figure/fig1-1.png)
Figure 1.1: The nitrogen cycle. The oxidation state of nitrogen in each intermediate is given in brackets. Denitrification is shown by black arrows, nitrogen fixation by a dark grey arrow and all other reactions by light grey arrows. Note that nitrification involves two reactions, oxidation of ammonia to nitrite and of nitrite to nitrate, but these have never been found together in the same organism. ANAMOX (anaerobic ammonia oxidation) is a poorly-characterised process in which nitrite and ammonia combine to give water and dinitrogen [1].
The cycle involves the interconversion between various oxidation states of nitrogen and is predominantly carried out by micro-organisms. A simplified view of the nitrogen cycle is one of a balance between two opposing processes. In nitrogen fixation atmospheric dinitrogen gas is reduced to ammonia, allowing the assimilation of nitrogen into biological molecules. Nitrogen is released back into the environment as ammonium during decay, oxidised to nitrate and converted to dinitrogen gas by the process of denitrification, the left hand branch of Figure 1.1. This involves the sequential reduction of nitrate to nitrite, nitric oxide, nitrous oxide and finally nitrogen, in a series of enzyme-catalysed reactions. The individual reactions of denitrification can be referred to as respiratory N-oxide reduction, because the process is used to generate ATP via a proton electrochemical gradient across the cytoplasmic membrane. The term dissimilatory N-oxide reduction is also used to indicate that the products are lost to the environment; this distinguishes the reactions from the assimilatory reductions used by plants and bacteria to incorporate nitrogen into biomass.
1.1.1 The importance of denitrification
Denitrification was first reported last century [2] and it has since been the focus of much research, spanning a variety of disciplines. Factors that affect the levels of N-oxides in the environment are of great ecological interest. For example, nitrous oxide, the penultimate compound in the pathway, has been implicated as a contributor to global warming and as an agent capable of destroying stratospheric ozone [3]. Denitrification is a route by which soil nitrogen may be lost, with implications for agricultural productivity [4]. The removal of nitrogenous compounds in waste water treatment by bioremediation is an important field, particularly in small countries such as the Netherlands [5], and the contribution of polluting nitrates in groundwater to eutrophication is another concern [6]. Nitrate and nitrite levels in the environment may also have health consequences, through conditions such as methaemoglobinaemia [7] and certain stomach cancers (caused by conversion of nitrites to N-nitroso compounds in the stomach) [8]. Information about microbial N-oxide metabolism may therefore be relevant to many researchers.
In recent years denitrifying bacteria have proven to be extremely interesting systems for the study of fundamental prokaryotic biochemistry, physiology and molecular genetics, and this aspect of denitrification is the main concern of this thesis. As a bioenergetic process, the enzymes involved may be used to investigate electron transfer and proton translocation, both of which are poorly understood at present. Biogenesis of the enzymes, in terms of targeting and compartmentalisation, folding and assembly of their various metal centres is another fundamental process. The catalytic mechanisms of each enzyme also attract research, particularly as several of the proteins involved are soluble, easy to purify in large amounts and, in some cases, have had their crystal structures determined. Finally there is always the possibility that study of prokaryotic systems may provide structural models for analogous eukaryotic processes. As an example, the bacterial cytochrome cd\(_1\) nitrite reductase (Section 1.5) generates bound nitric oxide which must be released from a haem group. In eukaryotes, nitric oxide participates in signal transduction by binding to a haem group on the enzyme guanyl cyclase, from which it must also be released [9].
1.2 The denitrifying micro-organisms
Denitrification research has traditionally focused on the Gram-negative bacteria (those containing a compartment between the cytoplasmic and outer membranes termed the periplasm) and until quite recently, respiratory N-oxide reduction was considered to be an exclusively prokaryotic attribute. This situation has altered somewhat with the discovery that at least two species of fungus, Fusarium oxysporum and Cylindrocarpon tonkinense can reduce nitrite to nitrous oxide [10,11]. In F. oxysporum, N-oxide reduction is localised to the mitochondrion [12] and is comprised of distinct enzymes for the dissimilatory reduction of nitrate [11], nitrite [13] and nitric oxide [14]. The system is significantly different to that found in bacteria, particularly the nitric oxide reductase which is a unique cytochrome P450-type enzyme.
The bacteria studied to date tend to fall into two groups; those in which at least one enzyme of the denitrification pathway has attracted interest, although the complete pathway has not been investigated (or may not be present), and those in which the complete denitrification pathway from nitrate through to dinitrogen has been characterised. Escherichia coli falls into the former category; although not a denitrifier, it possesses a membrane-bound nitrate reductase [15] and a putative periplasmic nitrate reductase [16], encoded by the nar and nap gene clusters, respectively, which are essentially identical to those found in the denitrifying bacteria. The genetic amenability of this organism and its wider importance in biochemical and genetic research has encouraged investigation of these enzymes; for example extensive sequencing of the E. coli genome allowed the identification of the E. coli nap gene cluster during a DNA database search using the Thiosphaera pantotropha nap genes [17].
Also in the first category are denitrifiers where one enzyme in the pathway has been singled out as being of specific interest. Examples include Achromobacter cycloclastes and Alcaligenes faecalis S-6; these organisms contain a copper nitrite reductase (Section 1.4.3), the X-ray crystal structures of which have been determined to high resolution [18,19]. This work has provided a great deal of information on copper centres and possible mechanisms of nitrite reduction.
However, the core of denitrification research concerns bacteria from the latter category; those in which the four enzymes required for complete denitrification have been identified and at least partially characterised. Many such species have been described [20] but the best studied are Pseudomonas aeruginosa, Pseudomonas stutzeri and Paracoccus denitrificans (and its close relatives, particularly T. pantotropha). P. denitrificans has long been of interest in the field of bioenergetics, due to the close similarity between its aerobic respiratory chain and that of the eukaryotic mitochondrion [21]. P. denitrificans has also been the subject of a great deal of metabolic research, due to its nutritional versatility. As well as growing anaerobically with nitrate as terminal electron acceptor, P. denitrificans can grow chemoautotrophically on inorganic substrates such as hydrogen or thiosulphate, chemoheterotrophically on organic acids such as acetate or succinate and methylotrophically, on methanol or methylamine. As with other bacteria denitrifying growth in P. denitrificans is an anaerobic process used as an alternative to aerobic respiration when oxygen, the preferred terminal electron acceptor, is in short supply.
1.3 Organisation of the denitrifying electron transport chain
Although denitrification is relatively widespread amongst the prokaryotes, detailed biochemical information is available for only a small number of organisms. However, the current model of the organisation of the denitrifying respiratory chain is shown in Figure 1.2.
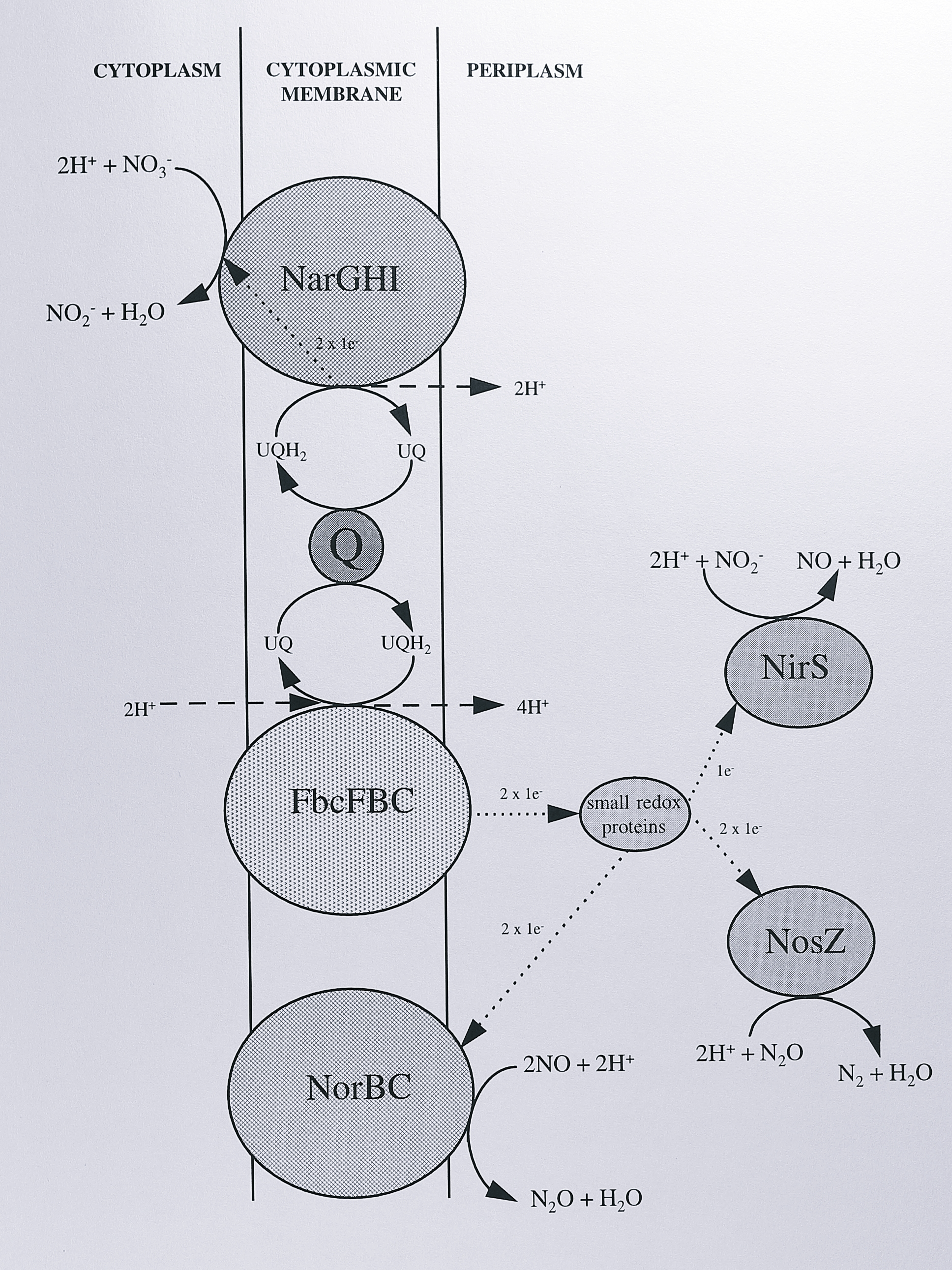
Figure 1.2: The denitrifying electron transport chain of Paracoccus denitrificans. The enzymes shown are: NarGHI, nitrate reductase, FbcFBC, cytochrome bc\(_1\) complex, NorBC, nitric oxide reductase, NirS, cyochrome cd\(_1\) nitrite reductase, NosZ, nitrous oxide reductase. ‘Q’ represents the ubiquinol/ubiquinone pool. Chemical reactions are indicated by solid arrows, proton translocation by dashed arrows and electron transfer by dotted arrows, with the stoichiometry shown in each case. Proton translocation at the cytochrome bc\(_1\) complex is described by the proton-motive Q-cycle (see text for details). Not shown is the periplasmic nitrate reductase, which is not present during anaerobic denitrifying growth conditions.
This diagram is based on the organism Paracoccus denitrificans, but is probably similar in other bacteria, particularly Ps. aeruginosa and Ps. stutzeri. During anaerobic denitrifying growth, a standard tricarboxylic acid cycle is employed, generating the reductants NADH and succinate. The cytoplasmic membrane thus contains prokaryotic homologues of the classical respiratory complexes: complex I (NADH-ubiquinone oxidoreductase), complex II (succinate-ubiquinone oxidoreductase) and complex III (ubiquinol-cytochrome c oxidoreductase), in addition to the integral membrane proteins nitrate and nitric oxide reductase. Nitrate reductase is believed to accept electrons from the ubiquinol pool. In contrast, the other three N-oxide reductases are believed to receive electrons from the cytochrome bc\(_1\) complex via small periplasmic electron transport proteins. Net proton translocation is achieved by the operation of the proton motive Q-cycle in the cytochrome bc\(_1\) complex [22] and the activity of Complex I. The reactions of N-oxide reduction are therefore periplasmic, with the exception of nitrate reduction which occurs in the cytoplasm [23]; this implies a transporter for entry of nitrate and export of nitrite to the periplasm (Section 1.4.1).
Denitrification is an energy conserving process. Consideration of the reduction potentials associated with each step (Table 1.1) suggests that each reaction is relatively exergonic, yielding sufficient free energy to drive the synthesis of ATP.
| Reaction | Couple | \(\Delta\)E\(^{o'}\) (V) | \(\Delta\)G\(^{o'}\) (kJ mol\(^{-1}\)) per reaction | \(\Delta\)G\(^o\) (kJ mol\(^{-1}\)) per NADH |
|---|---|---|---|---|
| \(\mathrm{2NO_3^- + 4H^+ + 4e^-}\) \(\rightarrow\) \(\mathrm{2NO_2^- + 2H_2O}\) | NO\(_3^-\)/NO\(_2^-\) | +0.420 | -285.6 | -142.8 |
| \(\mathrm{2NO_2^- + 2H^+ + 2e^-}\) \(\rightarrow\) \(\mathrm{2NO + 2H_2O}\) | NO\(_2^-\)/NO | +0.375 | -134.1 | -134.1 |
| \(\mathrm{2NO + 2H^+ + 2e^-}\) \(\rightarrow\) \(\mathrm{N_2O + H_2O}\) | NO/N\(_2\)O | +1.175 | -288.5 | -288.5 |
| \(\mathrm{N_2O + 2H^+ + 2e^-}\) \(\rightarrow\) \(\mathrm{N_2 + H_2O}\) | N\(_2\)O/N\(_2\) | +1.355 | -323.3 | -323.3 |
| Overall | ||||
| \(\mathrm{2NO_3^- + 2H^+ + 5H_2}\) \(\rightarrow\) \(\mathrm{N_2 + 6H_2O}\) | -1031.5 | -206.3 | ||
| \(\mathrm{O_2 + 4H^+ + 4e^-}\) \(\rightarrow\) \(\mathrm{2H_2O}\) | O\(_2\)/H\(_2\)O | +0.800 | -438.5 | -219.2 |
| \(\mathrm{NAD^+ + 2H^+ + 2e^-}\) \(\rightarrow\) \(\mathrm{NADH + H^+}\) | NAD\(^+\)/NADH | -0.320 |
With NADH as electron donor (E\(^o\)’ for the NADH/NAD\(^+\) couple is -320 mV), the redox span to nitric and nitrous oxide is actually greater than that for the oxygen/water couple. However, it must be born in mind that these values refer to standard state conditions and will be different in the cell; for instance the actual concentration of N\(_2\)O will be rather lower than one atmosphere. Furthermore, the spatial arrangement of the components involved in the electron transport sequence influences the stoichiometry of net proton translocation per pair of electrons passing through the electron transport chain. Termination of the denitrifying electron transport chain with nitrous oxide reductase results in no net proton translocation across the redox span from cytochrome c to nitrous oxide, whereas oxygen reduction by the cytochrome aa\(_3\) oxidase results in the equivalent of four protons translocated per pair of electrons. Thus, assuming a constant H\(^+\):ATP ratio for the ATP synthase, nitrous oxide respiration gives a lower ATP:2e\(^-\) ratio than does oxygen respiration. Nevertheless, denitrification represents an important alternative mode of respiration for those bacteria which have evolved the necessary enzymatic machinery, allowing them to exploit a wider range of environmental niches than obligate aerobes.
In the following sections, the enzymes of denitrification are discussed in more detail. The order of discussion is the same as that of each reaction in the pathway, with the exception of the cytochrome cd\(_1\) nitrite reductase; as this enzyme is the main focus of the project it is treated in greater detail after the others.
1.4 Enzymes of the denitrifying electron transport chain
1.4.1 The membrane-bound nitrate reductase
Membrane-bound nitrate reductase (Nar) is widely distributed, being found in many enteric bacteria as well as the denitrifying species. The most extensively characterised examples are from E. coli [24] and P. denitrificans [25]. Nitrate reductase from these sources is composed of three subunits with molecular masses of around 140 kDa (\(\alpha\)), 60 kDa (\(\beta\)) and 25 kDa (\(\gamma\)), as judged by SDS-PAGE. The \(\gamma\) subunit anchors the other two subunits to the cytoplasmic side of the cytoplasmic membrane and an \(\alpha\beta\) complex can be released from inside-out membrane vesicles by washing with EDTA or magnesium-free buffer [23]. Loss of the \(\gamma\) subunit during purification is accompanied by an apparent proteolytic cleavage at the C-terminus of the \(\beta\) subunit [26]. The inference from these studies is that the a\(\beta\) complex contacts the \(\gamma\) subunit through a region near the carboxy end of the \(\beta\) subunit polypeptide chain. Stoichiometry of the subunits is uncertain, but is thought most likely to be \(\alpha1\beta1\gamma1\); in E. coli the genes appear to have overlapping start and stop codons, which would result in translational coupling [27] and a stoichiometric synthesis of the subunits.
Membrane-bound nitrate reductase is a quinol oxidase, receiving from the quinol pool the two electrons required to reduce a molecule of nitrate. The electrons are transported through the complex by a variety of cofactors. The \(\gamma\) subunit contains two b-type haems [23]; modelling studies suggest that these conduct electrons from ubiquinol across the cytoplasmic membrane, leaving two protons in the periplasm in a classical “redox loop” mechanism [28]. The \(\alpha\beta\) complex contains several iron-sulphur clusters [29] and the \(\alpha\) subunit contains in addition a complex cofactor termed molybdopterin guanine dinucleotide (MGD) [30]. All bacterial nitrate reductases contain this cofactor, a variant of molybdopterin which in this case is conjugated to guanosine monophosphate. MGD-dependent enzymes form a large family, whose members include soluble and membrane-bound nitrate reductases, formate dehydrogenases and S-oxide reductases. Amino acid similarities between these sequences have been used to establish a number of sub-families and to identify conserved regions involved in the binding of metal cofactors [31].
A great deal of spectroscopic data has been amassed for the MGD cofactor, which receives electrons from the neighbouring \(\beta\) subunit, binds nitrate and catalyses an oxo-transfer reaction between nitrate and water [32]. EPR spectroscopy and mechanistic studies indicate that the molybdenum atom cycles between Mo(IV) and Mo(VI) states and is the site of the oxo-transfer reaction [33]. The complexity of the cofactor, in terms of the organic dithiolene moiety, is argued to provide a specific binding site for the Mo atom [34]. This has been confirmed by two recent crystal structures of the enzymes dimethyl sulphoxide reductase [35] and formate dehydrogenase H [36]. Both of these structures show that the molybdenum ion is co-ordinated by two MGD molecules through the dithiolene groups.
The structural genes for the three subunits of the complex, narGHI, have been cloned and sequenced from E. coli. A fourth gene, narJ, is found in this operon which encodes a cytoplasmic protein thought to be involved in biosynthesis of the complex [37]. This has been referred to as the \(\delta\) subunit but does not copurify with the enzyme. The T. pantotropha narJ and narHI genes have also been sequenced [28]. In E. coli, a second nitrate reductase operon, narZYWV, arising from gene duplication, is also found [38]. The E. coli narGHIJ operon is also linked to three other genes nar genes, narXL, a two-component nitrate/nitrite sensing regulator [39], Section 1.7.3 and narK, a putative nitrite exporter [40], or nitrate/nitrite antiporter [41]. Nitrate reduction is cytoplasmic; in E. coli the product nitrite may be reduced to ammonia by a cytoplasmic sirohaem-containing enzyme [42] or under some circumstances, exported to the periplasm by narK and reduced there by a c haem-containing nitrite reductase [43]. A similar system for nitrite export must operate in the denitrifying bacteria and has often assumed to be a nitrate/nitrite antiporter. No such protein has yet been identified in denitrifying bacteria, though other transport systems have been identified in prokaryotes In the cyanobacteria these include an active transporter of the ABC type [44] and a sodium-linked symporter [45], both of which may transport nitrate and nitrite, although nitrate reduction in these organisms is assimilatory rather than respiratory.
1.4.2 The periplasmic nitrate reductase
Periplasmic nitrate reductases have been purified from several bacteria, including R. capsulatus [46], T. pantotropha [47] and A. eutrophus [48]. In addition, the complete gene locus nap has been mapped and sequenced from T. pantotropha [16], R. sphaeroides [49] and E. coli [17]. Although purified from a relatively small number of species the enzyme is probably widespread amongst the Proteobacteria; probes for the gene have been used to isolate up to forty species of bacteria from soil and water environments in one locale [50].
The enzyme is a heterodimer of two subunits, NapA (90 kDa) and NapB (16 kDa) [47]. NapB is a dihaem cytochrome c\(_{552}\), which is believed to accept electrons from ubiquinol, via a quinol oxidase complex (possibly the napE gene product) and a tetrahaem cytochrome c (encoded by napC, [16]). NapA is the catalytic site, containing the MGD cofactor and, at least in T. pantotropha, an iron-sulphur cluster [51]. The enzyme is catalytically distinct from the membrane-bound nitrate reductase, in that it is not competitively inhibited by azide, nor can it reduce chlorate or bromate [47].
Extensive sequencing of the nap loci in T. pantotropha, R. sphaeroides and E. coli (the aeg-46.5 locus) has revealed a number of open reading frames besides those of the structural genes. T. pantotropha contains the aforementioned napE and napC, as well as napD, coding for a cytoplasmic protein of unknown function. E. coli contains napF, napG and napH; these proteins have been suggested to bind iron-sulphur clusters and to be involved in electron transport to NapAB [31]. In addition, there are a number of genes downstream of the nap cluster involved in c-type cytochrome biogenesis [52] and narP, a nitrate/nitrite response regulator (Section 1.7.3).
The physiological role of the periplasmic nitrate reductase is not especially clear and may even vary in different organisms. In T. pantotropha, the enzyme was initially assigned a role in aerobic denitrification, a feature that originally distinguished this organism from the very closely related P. denitrificans [53]. One rationale for this is that oxygen is known to inhibit nitrate transport to the cytoplasm [54], the site of reduction by the membrane-bound reductase, whereas a periplasmic location would effectively bypass this route of inhibition. However, more recent research seems to suggest that T. pantotropha does not denitrify aerobically to the extent previously reported [55], perhaps as a result of repeated sub-culturing in the laboratory. Furthermore, P. denitrificans also possesses a periplasmic nitrate reductase and synthesis of the other enzymes of denitrification appears to be quite strictly controlled by anaerobiosis. However, the periplasmic nitrate reductase may be expressed under conditions of stress (such as entering stationary phase, [48]) and as at least pre-synthesised cytochrome cd\(_1\) nitrite reductase appears to persist on switching from anaerobic to aerobic growth [56], the enzyme may play a role in aerobic denitrification as an adaptation to cope with rapidly changing levels of aeration in the environment.
Other roles have been suggested for the enzyme, involving the concepts of redox balance and redox poise. In photoheterotrophs and chemoheterotrophs, an optimum ratio of ubiquinol:ubiquinone is required to maintain turnover of enzyme complexes such as NADH dehydrogenase, the cytochrome bc\(_1\) complex and the photosynthetic reaction centre [31]. Certain growth conditions such as low light intensity or the provision of highly reduced carbon substrate can lead to an increase in the UQH\(_2\):UQ ratio and a fall in growth rate. The quinol oxidase activity of periplasmic nitrate reductase would alleviate this by consuming ubiquinol. There is good evidence for this model; in T. pantotropha, aerobic growth on the highly-reduced substrate butyrate results in a ten-fold increase in Nap expression compared to succinate [57], whilst nitrate reduction in photosynthetic R. capsulatus is markedly increased under low light intensity [58].
1.4.3 The copper nitrite reductase
Copper nitrite reductase is one of two types of eubacterial dissimilatory nitrite reductase, the other being the cytochrome cd\(_1\) enzyme discussed in Section 1.5. The two enzymes are distributed fairly evenly amongst bacteria with a slight preponderance of the cytochrome cd\(_1\) enzyme [59]. There is no correlation between genus (or even species) and the type of nitrite reductase present; for example Alcaligenes eutrophus H16 contains the cd\(_1\) type [60] as does A. faecalis strain IAM 1015 [61], whereas A. faecalis S-6 contains the copper enzyme [62]. The two enzymes are never found in the same organism but are functionally equivalent; expression of the copper nitrite reductase gene nirK in a Ps. stutzeri mutant lacking cytochrome cd\(_1\) restores nitrite reductase activity [63].
Copper nitrite reductases have been purified from several Gram-negative bacteria, including Achromobacter cycloclastes [64], Alcaligenes faecalis S-6 [62] and Pseudomonas aureofaciens [65] and one Gram-positive bacterium, Bacillus halodenitrificans [66]. It now seems clear that all the enzymes are homotrimeric, with each 37 kDa subunit containing a variant of a type I centre and one type II copper centre. Type I centres are mononuclear, cysteine-ligated, have high redox potentials and are an intense blue colour (but see below). They are always involved in electron transfer, rather than catalysis. Type II copper centres contain a single copper atom, with spectroscopic properties similar to those of inorganic copper complexes. They are ligated predominantly by histidine residues and are involved with catalysis at the active site, where substrate or exogenous ligands can displace a water-derived ligand. The type I centre in the copper nitrite reductase from A. cycloclastes is unusual in that it produces an intense green absorption at 458 nm; this is thought to be due to a short Cu-S-Met distance and substantial charge transfer through this bond [67].
Copper nitrite reductases are relatively well understood due to the elucidation of two X-ray crystal structures from the organisms A. cycloclastes [18] and A. faecalis S-6 [19]. These have revealed that each monomer forms two \(\beta\) barrel domains stabilised by a helical region. The histidine, cysteine and methionine ligands to the copper centres have also been identified; the type II copper centres are positioned at the subunit interfaces and share ligands from adjacent subunits. These centres can be removed and reconstituted, showing them to be the site of nitrite reduction. Some site directed mutagenesis of copper nitrite reductase has been carried out, which has identified the important role of the copper ligands [19], and the negatively-charged surface residues thought to interact with the electron donor pseudoazurin (Section 1.4.6 [68]). A mechanism of nitrite reduction has been proposed which is quite similar to the cytochrome cd\(_1\) enzyme (Section 1.5), involving protonation of the N-bound nitrite molecule by a neighbouring histidine residue, followed by dehydration to give a cuprous-NO+ species and followed by release of nitric oxide. The active site of copper nitrite reductase is comparable to that of carbonic anhydrase in that a number of similarly placed histidine residues act as proton donors [69]. However, there may be some mechanistic differences between the copper nitrite reductase and cytochrome cd\(_1\) -type nitrite reductase. No evidence for a Cu(I)-NO+ species has been obtained by H\({_2}^{18}\)O exchange experiments using the copper nitrite reductase, [70] and the enzyme also seems to produce significant amounts of N\(_2\)O as judged by isotope exchange experiments [71]. This occurs when nitric oxide levels are quite high, implying reaction between a labile Cu-NO species and NO or NO\(_2^-\). However, as with cytochrome cd\(_1\), this may not occur physiologically, as NO is normally rapidly removed in the cell by the activity of nitric oxide reductase (see below).
1.4.4 The nitric oxide reductase
For many years, it was unclear whether the product of nitrite reduction was nitric oxide or nitrous oxide and, if the former, whether nitric oxide was a free intermediate in denitrification. This situation has now been resolved by a number of methods: (1) trapping experiments using haemoglobin have shown nitric oxide to be freely diffusible [72], (2) membranes of mutants lacking nitrite reductase still possess a nitric oxide reductase activity [73] and most conclusively, (3) a distinct nitric oxide reductase enzyme has been identified, purified and shown to be essential for denitrification by gene disruption. Nitric oxide reductase has now been purified from several sources, including P. denitrificans [74], A. cycloclastes [75] and Ps. stutzeri [76]. On SDS-PAGE gels two subunits are seen; a cytochrome c of 17 kDa containing a single haem and a larger subunit (of apparent molecular mass 37 kDa, though actually 53 kDa from the gene sequence), binding a b-type haem. The enzyme also contains non-haem iron [76]. Biochemical studies of the protein solubilised in detergent vesicles have been carried out; cytochrome c\(_{550}\) can donate electrons to the enzyme and the K\(_m\) for nitric oxide under these conditions is sub-micromolar [74]. This measurement seems reasonable as NO concentrations in whole cells are generally 10\(^{-7}\) M or less [77].
Redox titrations have revealed only two redox components in the enzyme; the c haem component (+225 mV in P. denitrificans, +280 mV in Ps. stutzeri) and the b haem component (+305 mV or +322 mV in the same organisms respectively, Chan and Ferguson, unpublished work, [78]). Early studies reported a stoichiometry of two haems per NorBC complex [76], but more recent work indicates that there are two b-type haems (one high-spin that can bind carbon monoxide, the other low-spin), one c haem and one non-haem iron per enzyme complex [79,80]. The membrane-bound nature of the enzyme makes purification quite difficult and it now seems that the earlier estimates of haem stoichiometry were erroneous.
The problems outlined above have made elucidation of a reaction mechanism for nitric oxide reductase quite problematic; in particular it was difficult to envisage how dimerisation of two NO molecules could be achieved at a single haem binding site. However, the cloning and sequencing of the norBC genes has led to several suggestions for a catalytic mechanism. The NorB subunit has structural similarity to subunit I of cytochrome c oxidase [81], in that both are predicted to have 12 transmembrane helices, with 6 conserved histidines that act as ligands (in the oxidase, to haems a and a\(_3\) and to the Cu\(_{A3}\) site, [82]. Furthermore, like nitric oxide reductase the more primitive cytochrome oxidase, cytochrome cbb\(_3\), has a subunit II (FixN) possessing a cytochrome c, rather than a binuclear Cu\(_A\) centre [83]. These analogies have strengthened the case for two b-type haems and the conserved histidine residues strongly suggest liganding to the non-haem iron identified in later preparations of nitric oxide reductase. This raises the intriguing possibility of NO reduction at a binuclear haem-non-haem iron centre. Modelling of this enzyme family has also been used to suggest an evolutionary progression from nitric oxide reductase, through cytochrome cbb\(_3\) to the cytochrome c oxidases, and to imply that the cytochromes cbb\(_3\) may have a different mechanism of proton pumping to the haem-copper enzymes, whilst nitric oxide reductase is probably not a proton pump [81]. It is notable that cytochrome c oxidase is capable of nitric oxide reduction [84] and a recent paper describing nitric oxide reductase from T. pantotropha indicates that it has a cytochrome oxidase activity [79].
1.4.5 The nitrous oxide reductase
Nitrous oxide reductase, the final enzyme of the denitrification pathway is one of the most comprehensively studied of the denitrifying enzymes. It has been purified from numerous sources, including Ps. stutzeri [85], P. denitrificans [86], Ps. aeruginosa [87] and A. faecalis [88]. The enzyme appears essentially identical from all sources, being a homodimer of subunits each with a molecular mass of around 70 kDa. Nitrous oxide reductase is a soluble, periplasmic enzyme and contains four copper atoms per polypeptide [85]. Spectroscopic models indicate that two types of site are present; a Cu\(_A\) site (sometimes termed the “purple copper centre” due to its distinctive colour), related to that found in cytochrome c oxidase and which functions in electron transfer, and a catalytic site, variously termed Cu\(_B\), Cu\(_C\) or Cu\(_Z\), the latter term being preferred. Of these the Cu\(_A\) site is the better understood and has been extensively modelled on the basis of EPR and MCD data. It is similar to the site found in cytochrome c oxidase [89]; sequence similarity between the C-terminal of nitrous oxide reductase and cytochrome c oxidase subunit II (which binds Cu\(_A\)) has been used to predict the ligands to the nitrous oxide reductase copper centre [90]. In general, these predictions are supported by spectroscopic studies. The EPR spectrum of this centre is quite unusual and suggests that one electron is delocalised over both copper atoms, making the centre a one-electron acceptor [91]. The catalytic Cu\(_Z\) site is rather less well understood, largely because it has fewer distinct spectroscopic properties. It has also been modelled by [91] and it is also thought to be dinuclear, containing 2 Cu(II) atoms. Reduction using dithionite is thought to reduce only one of these atoms to Cu(I), giving a dead-end product which is enzymically inactive. Ligands to the Cu\(_Z\) site have been suggested by examining conserved residues known not to be involved with the Cu\(_A\) site; 8 histidines and a single cysteine are possible candidates.
The enzymology of nitrous oxide reductase is rather complex. Two forms of the protein can be purified; the purple form (isolated anaerobically) and the pink form (isolated aerobically), with distinct spectroscopic features [92], the anaerobic form having a higher specific activity. Reduction of the enzyme with dithionite followed by oxidation by molecular oxygen converts the purple form to the pink form. Dithionite reduction alone produces a third form of the enzyme, the “blue” form which still contains oxidised copper; this is associated with the Cu\(_Z\) centre. The enzyme is inhibited by acetylene, azide, carbon monoxide and cyanide [93], which presumably can act as copper ligands at one or both of the copper centres.
The purified enzyme shows the highest activities when assayed with reduced viologens as electron donors, activities using the presumed physiological donors are lower. However, turnover-dependent inactivation has been noted for some of these donors e.g. [94]. The activity of the enzyme from some species has been increased by incubation with reduced viologens [86] and also by exposure to high pH buffer [85]; the latter induces a permanent chemical change and it has been suggested that this reaction converts inactive forms of the Cu\(_Z\) site to active species, perhaps by regenerating a thiolate ion [31].
The complexity of the copper sites in nitrous oxide reductase has slowed progress in the understanding of its mechanism. However, the dinuclear nature of the Cu\(_Z\) site suggests that nitrous oxide is bound as a bridging ligand between the two copper atoms; following electron transfer from the Cu\(_A\) site, the N-O bond could be activated for cleavage, releasing dinitrogen. The intermolecular electron transfer is another intriguing aspect of this enzyme, in particular why the unusual purple copper centre is used in preference to the more normal type I blue copper centre. The electron pathway through the enzyme (whether through hydrogen bonds/through space or through covalent bonds) is another problem that remains to be elucidated.
The structural gene for nitrous oxide reductase, nosZ, has been cloned and sequenced from a number of sources, allowing the sequence analyses described previously. In Ps. stutzeri [95], it is part of a large denitrification cluster containing a number of genes believed to be involved in biosynthesis of the enzyme. Similar clusters are thought to exist in Ps. aeruginosa [90] and P. denitrificans [96], though there is no evidence of the accessory genes in A. eutrophus [90]. The nos gene cluster is discussed more fully in Section 1.6, in relation to the other genes of denitrification. An interesting feature of the nosZ genes is that the encoded NosZ protein appears to have an unusually long periplasmic targeting sequence (58-60 amino acids), compared to the more usual 26 or less [97]. Another unusual feature is that the N-termini of the targeting sequences contain a quite highly conserved motif; these features have been suggested to point to a common export pathway for periplasmic proteins with complex metal cofactors [98].
1.4.6 Small electron transfer proteins
The periplasms of Gram-negative bacteria contain a variety of small, soluble redox proteins. These are assumed to mediate electron transfer between membrane-bound redox complexes, such as the cytochrome bc\(_1\) complex and the periplasmic enzymes, including those of denitrification, although in few cases has an in vivo reaction been directly demonstrated. The small electron transfer proteins group into two types: the small cytochromes c and the cupredoxins.
The cytochromes c are a large family of proteins, that have been grouped into various families by consideration of size, sequence similarity and physicochemical properties [99]. They have been popular subjects for the study of protein structure and function for many years and a vast literature exists on the subject, which can only be briefly summarised here. The periplasmic cytochromes c involved in electron transfer include cytochrome c\(_{550}\) from P. denitrificans, cytochrome c\(_{551}\) from Ps. aeruginosa and cytochrome c\(_2\) from Rhodobacter capsulatus. These proteins are characterised as Class I cytochromes c: they are generally small (80-120 amino acids), and contain one covalently bound c haem. The haem group is low-spin, co-ordinated by histidine and methionine and found near the N terminus of the protein. Many crystal structures have been determined, revealing a mostly a helical structure, a crevice in which the haem group is buried and a positively-charged face around the haem edge, through which redox partners may interact by electrostatic interactions [100].
The cupredoxins are a family of small (around 13 kDa) copper-containing proteins containing what are known as type I or “blue copper” centres. They contain a single copper atom which is involved in electron transfer with donor and acceptor proteins. Cupredoxins absorb intensely near 600 nm giving them an intense blue colour. The copper atom is liganded by two histidine residues, a cysteine and a weak methionine ligand; subtle variations in the ligand geometry give rise to four spectroscopic sub-classes of cupredoxin: (i) azurins, (ii) pseudoazurins, (iii) plastocyanin/amicyanin, (iv) stellacyanin and associated proteins. Examples of cupredoxins include pseudoazurin from T. pantotropha [101], azurin from Ps. aeruginosa [102] and amicyanin from P. denitrificans [103]. A number of X-ray crystal structures are known which show a high degree of similarity; cupredoxins contain a Greek key \(\beta\)-barrel fold, consisting of 8 \(\beta\) sheets. As with the cytochromes c, surface regions that govern the interactions between the cupredoxins and their redox partners have been defined; in azurin, His-117 is thought to transfer electrons to partners bound by a patch of hydrophobic residues around this residue [104]. However, recent analysis of several redox partners has introduced the concept of pseudospecific interactions, in which both hydrophobic patches and oppositely charged surface regions act to bring proteins together and promote electron transfer [105].
Proteins are generally assumed to be physiological redox partners on the basis of their reactivity in in vitro assays, as it is difficult to prove an in vivo relationship. Where this has been attempted, mutant strains lacking the small redox protein under study have been used. A mutant of P. denitrificans lacking cytochrome c\(_{550}\) has been shown to denitrify at a similar rate to the wild-type organism but anaerobic growth becomes sensitive to diethyldithiocarbamate (a copper chelator), suggesting a role for a copper protein (probably pseudoazurin) in electron transport [106]. It is often the case that other redox proteins can substitute for an absent protein in redox carrier mutants; for example a mutant of R. capsulatus lacking cytochrome c\(_2\) can still grow photosynthetically, because a membrane-bound cytochrome c takes over its role [107].
Redox partners may also be inferred from co-expression. In P. denitrificans grown methylotrophically, two additional cytochromes (c\(_{551i}\) and c\(_{553i}\), [108]) and the copper protein amicyanin [103] appear, suggesting that they are involved in electron transfer to or from the methanol dehydrogenase.
1.5 The cytochrome cd\(_1\) nitrite reductase
Cytochrome cd\(_1\) nitrite reductase is the alternative dissimilatory nitrite reductase to the copper enzyme (Section 1.4.3). Cytochrome cd\(_1\) was first isolated from Ps. aeruginosa [109]. It reduces oxygen and has been referred to as soluble cytochrome oxidase, but it was later shown that the physiological role of the enzyme is nitrite reduction. This was based on the findings that the K\(_m\) for nitrite is much lower than that for oxygen and that the enzyme is induced during anaerobic growth on nitrate [110]. However oxygen reduction still attracts interest; it is known to be a four-electron reduction to water [111] and may still prove relevant in the study of membrane bound cytochrome oxidases.
Cytochrome cd\(_1\) has since been identified, isolated and studied in detail from many sources; some of these include P. denitrificans [112], T. pantotropha [101], Ps. stutzeri [73], A. faecalis [61], A. eutrophus [60], Thiobacillus denitrificans [113], Magnetospirillum magnetotacticum [114] and Roseobacter denitrificans (formerly Erythrobacter sp. Och114 [115]). Their gross properties are all similar, though as will be discussed later, there are important differences between them. The enzyme from Ps. aeruginosa is the most intensively studied of the family in terms of biochemical mechanistic data and most of the discussion below pertains to data from this enzyme.
1.5.1 Subunit structure
Cytochrome cd\(_1\) from all sources is a dimer of two identical subunits of molecular masses around 55-65 kDa, as judged by SDS-PAGE, size exclusion chromatography and sedimentation analysis [116]. Each subunit contains two haem groups; a covalently-bound c haem identical to that found in mitochondrial cytochrome c and a non-covalently bound d\(_1\) haem, which is unique to this enzyme (Figure 1.3).
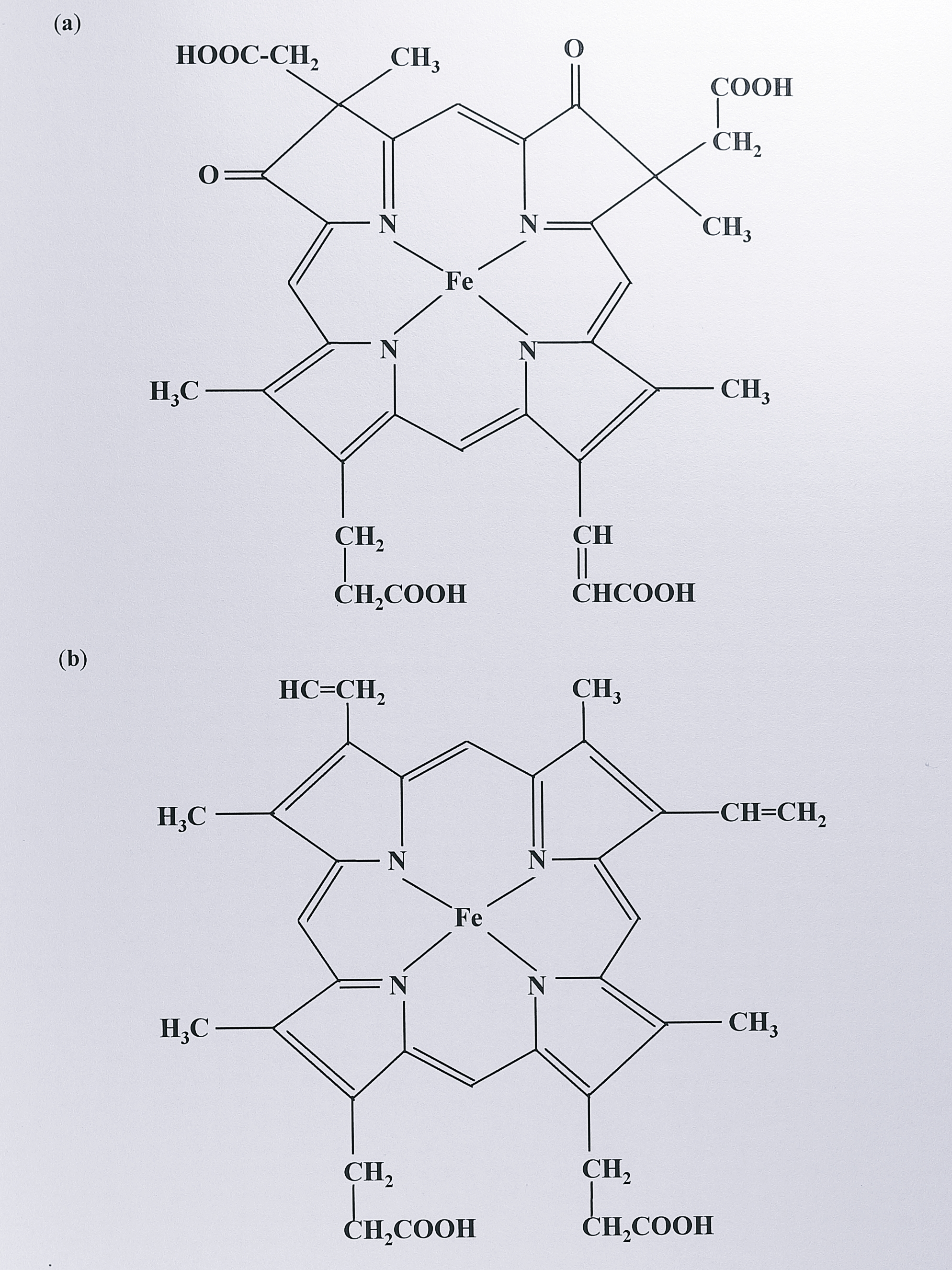
Figure 1.3: (a) Structure of d\(_1\) haem and (b) of protoporphyrin IX. The d\(_1\) haem is unique to cytochrome cd\(_1\). Protoporphyrin IX can occur as a non-covalently bound prosthetic group, where it is referred to as b-haem. In c-type cytochromes, protoporphyrin IX binds covalently via the vinyl side chains to two cysteine residues in the polypeptide at the consensus site CXXCH (X = any amino acid).
The latter haem is distinguished by the presence of oxo and carboxylate moieties and partial saturation on two adjacent pyrroles (I and II) of the porphyrin ring. Its structure has been confirmed by NMR spectroscopy and total chemical synthesis [117]. Haem d\(_1\) can be removed from the protein to give an inactive semi-apo protein; activity can be restored by reconstitution with the haem group, or to a much lesser extent, a-type haem [118].
1.5.2 Properties of the haem groups in cytochrome cd\(_1\)
Haem groups and their ligands are fundamental determinants of the catalytic properties of enzymes like cytochrome cd\(_1\). The ligands to the haems are amino acid residues from the polypeptide chain; they control access of the substrate and the electrochemistry of the reaction by modulating the reduction potential of the haem. Cytochrome cd\(_1\) has been investigated by numerous spectroscopic methods, including UV-visible spectroscopy, nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR) and magnetic circular dichroism (MCD), from which models have been developed to explain the observed spin states and absorption features of the haem groups. In addition, a number of haem redox potentials have been measured for the enzyme.
In fully oxidised cytochrome cd\(_1\) both haems are in a low spin state, indicating that they are hexacoordinate. Reduction causes the d\(_1\) haem to become high spin indicating that it has lost an axial ligand [119]. This implies that the d\(_1\) haem is the site of nitrite binding and a number of ligands have been used to probe this binding-site, including azide, imidazole [120], cyanide [121] and carbon monoxide [122]. Similar behaviour has been noted for the periplasmic cytochrome c peroxidase enzyme of P. denitrificans, in which peroxide access at the catalytic site is thought to be blocked by a sixth axial ligand until the electron receiving haem group is fully reduced; this would prevent partial reduction of hydrogen peroxide to more reactive oxygen species [123].
The identity of the ligands to the haems has taken a long time to resolve and apparently conflicting results have been reported for different cytochromes cd\(_1\). Low temperature MCD and EPR spectra of the Ps. aeruginosa enzyme in the oxidised state suggest histidine-methionine ligation at the c-haem and possibly bis-histidine ligation at the d\(_1\) haem [124]. This is also the case for Ps. stutzeri [125]. In contrast, EPR and Mössbauer spectroscopy of the enzyme from Th. denitrificans suggested bis-histidine ligation at both haems [126], whilst the T. pantotropha crystal structure (Section 1.5.3.1) has bis-histidine at the c haem and an unusual histidine-tyrosine at the d\(_1\) haem [127]. Midpoint redox potentials of the haems have been reported for Ps. aeruginosa by several groups, with some disagreement. The values reported to date are summarised in Table 1.2.
| Organism | E\(^{o'}\) of c haem (mV) | E\(^{o'}\) of d\(_1\) haem (mV) | E\(^{o'}\) (c - d\(_1\)) (mV) | Reference |
|---|---|---|---|---|
| Pseudomonas aeruginosa | 73 | Horio et al. (1961) [128] | ||
| 288 | Kamen and Horio (1970) [129] | |||
| 290 | 40-100 | Wharton et al. (1973) [130] | ||
| 24 | Shimada and Orii (1976) [131] | |||
| 60 | Blatt and Pecht (1979) [132] | |||
| 294 | 287 | Schichman and Gray (1981) [133] | ||
| 288 | Silvestrini et al. (1982) [134] | |||
| 25 (pH 8) | Carson et al. (1986) [135] | |||
| Pseudomonas nautica 617 | 234 (pH 7.6) | 199 (pH 7.6) | Besson et al. (1995) [136] | |
| Magnetospirillum magnetotacticum | 191 | 180 | Yamazaki et al. (1995) [114] |
Measurement seems to be complicated by a strong cooperative redox reaction between the haems [132] and by the possibility of d\(_1\) haem loss and protein denaturation during the experiment (Alrik Koppenhöfer, personal communication). The c haem value seems the more consistent of the two; for Ps. aeruginosa this is estimated at around +288 mV (pH 7.0). The d\(_1\) value has been extrapolated from data used to measure the difference between the two haems and has been variously estimated as being between 25 and 70 mV lower. For the enzyme from M. magnetotacticum (which may be an atypical member of the family), the c and d\(_1\) values are estimated as +191 and +180 mV respectively [114]. The corresponding values reported for Pseudomonas nautica strain 617 cytochrome cd\(_1\) are +234 mV and +199 mV (pH 7.6) [136]. The c haem in the semi-apo form of T. pantotropha cytochrome cd\(_1\) has been estimated as +243 mV (Koppenhöfer and Ferguson, unpublished work). The values are in the expected range given that the c haem is expected to receive electrons from donors of potentials around +250 mV, though the d\(_1\) values are somewhat lower than expected given that electrons are thought to flow from the c to the d\(_1\) haem.
1.5.3 Folding and conformation of cytochrome cd\(_1\)
At present seven primary sequences of cytochromes cd\(_1\) are known, from the organisms Pseudomonas aeruginosa [137], Pseudomonas stutzeri Zobell [138], Pseudomonas stutzeri JM300 [139], Paracoccus denitrificans IFO 12442 [140], Paracoccus denitrificans PD1222 [141], Alcaligenes eutrophus H16 [142] and Thiosphaera pantotropha (this work, Chapter 3). The first to be obtained was from Ps. aeruginosa and secondary structure predictions for this protein indicated that it contained two domains; a predominantly \(\alpha\) helical N-terminal portion (including the c-haem region) and folds of largely \(\beta\)-sheet towards the carboxy terminus [137]. Further evidence of a two domain structure was obtained from partial digests with the alkaline serine protease subtilisin, which indicated that a dimer containing only d\(_1\) haem could be obtained [143]. Circular dichroism spectroscopy using the Ps. aeruginosa enzyme indicated that the proportions of \(\alpha\) helix and \(\beta\)-sheet in the oxidised enzyme were 16% and 48% respectively, with a change on reduction to 35% \(\beta\)-sheet [144].
Conformational changes have been observed when the enzyme is reduced, although the precise character of these changes is unknown. In addition to a decrease in \(\beta\)-sheet content the radius of gyration decreases as measured by small angle X-ray scattering [145], the thermal stability of the protein decreases [146] and crystals of cytochrome cd\(_1\) crack on reduction [147]. These observations point to some kind of rearrangement in quaternary structure with a change in redox state.
The orientation and relative position of the haem groups has long been a contentious issue. A slow electron transfer rate between the haems as observed by some groups suggested that they may be quite far apart. These data are contradicted by fluorescence quenching studies which indicated that the haems were grouped at one end of a subunit [148] and by the recently obtained structure of the T. pantotropha enzyme (Section 1.5.3.1). In the latter case, the haems are at a relative angle of about 60\(\unicode{x00b0}\) and the iron-iron distance between haems within a subunit is 20.6 \(\unicode{x212b}\) [127].
1.5.3.1 Crystal structure of the T. pantotropha enzyme
Cytochrome cd\(_1\) from Ps. aeruginosa was first isolated over thirty years ago. Crystallisation of the enzyme was reported almost immediately [149] but although other groups have also obtained crystals [147,150], none of this work has led to a published X-ray structure. However, the crystal structure of cytochrome cd\(_1\) in the oxidised state from T. pantotropha has recently been determined (Figure 1.4).
![Crystal structure of cytochrome _cd_$_1$ in the oxidised form from _T. pantotropha_. Taken from Fülöp _et al_. (1995) [@fulop_anatomy_1995]. (a) shows the dimeric molecule with the $\alpha$-helical _c_-haem domain at the top and the $\beta$-propeller _d_$_1$ haem domain below. (b) shows the _d_$_1$ haem domains of each subunit from below, clearly illustrating how the eight-fold $\beta$-propeller structure forms a channel to bind the _d_$_1$ haem. The chain of each subunit is coloured from blue (N-terminus) to red (C-terminus).](figure/fig1-4.png)
Figure 1.4: Crystal structure of cytochrome cd\(_1\) in the oxidised form from T. pantotropha. Taken from Fülöp et al. (1995) [127]. (a) shows the dimeric molecule with the \(\alpha\)-helical c-haem domain at the top and the \(\beta\)-propeller d\(_1\) haem domain below. (b) shows the d\(_1\) haem domains of each subunit from below, clearly illustrating how the eight-fold \(\beta\)-propeller structure forms a channel to bind the d\(_1\) haem. The chain of each subunit is coloured from blue (N-terminus) to red (C-terminus).
The T. pantotropha structure was initially determined at a resolution of 1.55 \(\unicode{x212b}\) from diffraction data and using the primary sequence of the P. denitrificans PD1222 protein as a guide [127]. The latter sequence was assumed to be highly homologous to that of the protein from T. pantotropha and this has been shown subsequently to be the case (Chapter 3).
The crystal space group contains a dimer with each monomer organised in two domains, as predicted by earlier studies. The smaller domain is predominantly \(\alpha\)-helical, binds the c haem and resembles a Class I fold to some degree, though with some significant differences. The ligands to this haem are His-17 and His-69; this is rather unexpected as bis-histidine ligation is normally associated with low redox potential [151], whereas the c haem is thought to have a redox potential in the +250 mV range. The larger domain is an eight-bladed \(\beta\)-propeller structure, a fold previously identified in methanol dehydrogenase [152]. Propeller structures with fewer blades are also seen in methylamine dehydrogenase (seven blades [153]) and neuraminidase (six blades [154]). The central channel of this structure contains the d\(_1\) haem, which is ligated by His-200 and interestingly, by Tyr-25. Not only is this an unusual haem ligand, but it originates in the c haem domain; hence although separate, there is a distinct structural link between the domains. There are also a large number of important hydrogen bonds at the domain interface, and between subunits.
The structure answers some of the earlier questions about haem position and environment. The two haem types are at an angle of about 60\(\unicode{x00b0}\) to each other within a monomer and separated by an iron-iron distance of 20.6 \(\unicode{x212b}\). The haem edge-edge distance in a monomer is 11.0 \(\unicode{x212b}\). Haem distances between subunits are even larger; c haem iron-iron is 42.4 \(\unicode{x212b}\). c haem-d\(_1\) haem is 47.0 \(\unicode{x212b}\) and d\(_1\) haem iron-iron is 50.5 \(\unicode{x212b}\), with no edges closer than 38.5 \(\unicode{x212b}\). This would seem to rule out a simple iron-iron electron transfer mechanism and also discounts a binuclear haem binding site for oxygen reduction. The reported crystal structure, in combination with more recent work on reduced crystals of the enzyme and previous mechanistic studies, has allowed the development of a model for nitrite reduction which is discussed in the next section.
1.5.4 Kinetic and mechanistic studies of the enzyme
The mechanism of nitrite reduction and electron transfer in cytochrome cd\(_1\) has been investigated by a number of methods. Classical enzymological studies have been limited by the fact that the product of nitrite reduction, nitric oxide, is a potent inhibitor of the enzyme and may also bind to the haem groups of donor proteins used in the assay system; this leads to rather complex patterns of product inhibition and rate equations [155,156]. However, a considerable amount of information has been gained from isotopic labelling techniques, spectroscopy and fast kinetic methods and data so obtained, combined with structural information, have led to a coherent model for nitrite reduction (Figure 1.5).
![Proposed mechanism for nitrite reduction by cytochrome _cd_$_1$ from _Thiosphaera pantotropha_ (Fülöp _et al_., 1995 [@fulop_anatomy_1995]). (a) Nitrite binds at the reduced _d_$_1$ haem which is pentacoordinate, displacing a water molecule. (b) His-345 and His-388 act as proton donors, allowing the abstraction of an oxygen atom from bound nitrite. (c) This leaves nitric oxide bound at the oxidised _d_$_1$ haem. (d) Bound NO is then displaced by replacement with Tyr-25. The _d_$_1$ haem is re-reduced by an electron from the _c_-haem, displacing Tyr-25 and allowing the cycle to begin again.](figure/fig1-5.png)
Figure 1.5: Proposed mechanism for nitrite reduction by cytochrome cd\(_1\) from Thiosphaera pantotropha (Fülöp et al., 1995 [127]). (a) Nitrite binds at the reduced d\(_1\) haem which is pentacoordinate, displacing a water molecule. (b) His-345 and His-388 act as proton donors, allowing the abstraction of an oxygen atom from bound nitrite. (c) This leaves nitric oxide bound at the oxidised d\(_1\) haem. (d) Bound NO is then displaced by replacement with Tyr-25. The d\(_1\) haem is re-reduced by an electron from the c-haem, displacing Tyr-25 and allowing the cycle to begin again.
Electrons from donor proteins are believed to enter the enzyme at the c haem [157]. This is followed by an intramolecular electron transfer to the d\(_1\) haem. Reduction of this haem causes the loss of one of its axial ligands, allowing productive binding of nitrite. Isotope exchange studies have shown that the nitrite ion is protonated, then dehydrated to give an enzyme-bound nitrosyl intermediate [158]. Some earlier exchange studies also indicated that a substantial amount of reaction product was nitrous oxide [159]; however this has now been discounted as an artefact of the assay system, as P. denitrificans cytochrome cd\(_1\) assayed using isoascorbate as electron donor generates stoichiometric amounts of nitric oxide from nitrite [160]. A bound Fe(III)-NO+ intermediate would not be detectable by EPR, but has recently been observed by FTIR spectroscopy [161]. Furthermore, stopped-flow kinetic studies show that the first observable species is enzyme-bound nitric oxide at the oxidised d\(_1\) haem, with the c haem reduced [162]. It is notable that in this assay system, a slow intramolecular electron transfer from the c haem to the d\(_1\) haem occurs, giving fully-reduced NO-bound enzyme as the final product. Bound NO has also been observed by EPR [163]. This implies that the protonation/dehydration step is very fast and is followed by rapid oxidation of the d\(_1\) haem. The c haem is also re-reduced rapidly by a second electron; this rate is increased by the presence of NO at the d\(_1\) haem [162].
In T. pantotropha cytochrome cd\(_1\), Tyr-25 is proposed to be the axial ligand which leaves the d\(_1\) haem on reduction. This ligand is then proposed to “swing back”, displacing the nitric oxide molecule from the oxidised d\(_1\) haem. Such a scheme would mean that NO had to be displaced before the d\(_1\) haem could be reduced by the second electron from the c haem (in contrast to [162]); this makes mechanistic sense as normally, Fe(II)-NO complexes are very stable, with a half-life for dissociation of the NO in the region of days [164]. This could still allow for the slow intramolecular electron transfer rate observed in some stop-flow experiments which has been assigned as the rate-limiting step [162]; the electron would have to “wait” at the c haem for ligand reorganisation around the haem d\(_1\) co-ordination sphere. However, the observed intramolecular transfer rate may vary according to the precise experimental method; more recent work using the technique of pulse radiolysis suggests that it may not be as slow as was previously thought [165].
Although the scheme outlined above is very plausible and well-supported by evidence, there are still several issues to be resolved. One of these is the precise nature of the enzyme cycle; recently it has been shown that on reduction, the c haem of cytochrome cd\(_1\) from T. pantotropha changes from bis-histidine to histidine-methionine ligation [166]. Thus it may be that the fully-oxidised form of the enzyme is an artefact of purification and that during the catalytic cycle in vivo, the completely oxidised state is not regenerated. Another problem is relating mechanistic data concerning the enzyme from different sources to the crystal structure of the enzyme from T. pantotropha, particularly as mounting spectroscopic evidence indicates that axial haem ligation does seem genuinely to differ between cytochrome cd\(_1\) enzymes from different species; this point is discussed more fully in Chapter 3.
1.6 Genetic analyses of denitrification
As outlined in the previous sections, a large number of genes involved in denitrification have been mapped and sequenced from several bacterial species. The first of these was the structural gene for nitrous oxide reductase, nosZ, from Ps. stutzeri Zobell; the gene was isolated using Tn5 transposon mutagenesis and analysis of mutants deficient in denitrifying growth [167]. Subsequent cloning and sequencing of the flanking DNA led to the discovery of a large cluster of over 30 denitrification genes in this organism, spanning over 30 kb [95]. Other workers have built on this approach by screening digested genomic DNA in different organisms using oligonucleotide probes for specific denitrification genes, isolating large hybridising fragments and sequencing. In this way, similar denitrification gene clusters have been identified in Ps. aeruginosa and P. denitrificans. Maps of the largest contiguous clusters as presently known are shown in Figure 1.6.
![Denitrification gene clusters in _Ps. stutzeri_ Zobell, _Ps. aeruginosa_ and _P. denitrificans_ PD1222. Loci within the clusters that contain genes for nitrite- (_nir_), nitric oxide- (_nor_) and nitrous oxide- (_nos_) reduction are indicated. Genes in these regions are labelled with single letters. Numbered genes are open reading frames that have not been designated a gene name. Homologous genes that are found in all three clusters are indicated by identical shading; the structural genes for nitrite reductase (_nirS_) and nitric oxide reductase (_norCB_) are shaded black. Note that a gene downstream of _norD_ designated _fnrD_ in _Ps. stutzeri_ Zobell is thought to be a homologue of _dnr_ (_Ps. aeruginosa_) and _nnr_ (_P. denitrificans_), Zumft and Körner (1997) [@zumft_enzyme_1997] but its sequence has not yet been reported. The figure was compiled from sequence data contained in the following GenBank accession numbers: _Ps. stutzeri_ Zobell Z69589, 250198, X56813, X53676, Z13988, S41769, X65277, Z17423, S51580, Z73914, Z28384; _Ps. aeruginosa_ D50473, D38133, D37883, X16452, D50019, D84475; _P. denitrificans_ U28078, U05002, U17435. Additional data for P. denitrificans was derived from de Boer (1996) [@de_boer_regulation_1996].](figure/fig1-6.png)
Figure 1.6: Denitrification gene clusters in Ps. stutzeri Zobell, Ps. aeruginosa and P. denitrificans PD1222. Loci within the clusters that contain genes for nitrite- (nir), nitric oxide- (nor) and nitrous oxide- (nos) reduction are indicated. Genes in these regions are labelled with single letters. Numbered genes are open reading frames that have not been designated a gene name. Homologous genes that are found in all three clusters are indicated by identical shading; the structural genes for nitrite reductase (nirS) and nitric oxide reductase (norCB) are shaded black. Note that a gene downstream of norD designated fnrD in Ps. stutzeri Zobell is thought to be a homologue of dnr (Ps. aeruginosa) and nnr (P. denitrificans), Zumft and Körner (1997) [168] but its sequence has not yet been reported. The figure was compiled from sequence data contained in the following GenBank accession numbers: Ps. stutzeri Zobell Z69589, 250198, X56813, X53676, Z13988, S41769, X65277, Z17423, S51580, Z73914, Z28384; Ps. aeruginosa D50473, D38133, D37883, X16452, D50019, D84475; P. denitrificans U28078, U05002, U17435. Additional data for P. denitrificans was derived from de Boer (1996) [169].
Knowledge of these gene sequences has allowed analysis of the putative gene products using a range of methods. Some of these are theoretical; for instance the sequence can be analysed for known motifs (such as the CXXCH c haem binding motif), or subjected to computer-based methods which may predict properties such as sub-cellular location, the presence of a periplasmic targeting sequence or the number of transmembrane helices in a membrane protein. Database searching may also reveal possible homologues whose function is already known. All of these methods have been applied to the genes of denitrification.
In general, experimental verification of gene function lags behind the sequencing effort. However, some progress has been made in this area. The commonest approach is to inactivate the gene of interest, usually by inserting an antibiotic resistance cassette into the gene by homologous recombination, and then to look for phenotypic effects under denitrifying growth conditions. This approach has the disadvantage that it may have polar effects on genes downstream of the target gene, but such effects can normally be recognised by complementation experiments. Using the methods outlined above, a number of similarities in the denitrification gene clusters of P. denitrificans, Ps. stutzeri and Ps. aeruginosa are apparent, that can be summarised as follows.
All three clusters contain the structural genes for nitric oxide reductase, norCB, and nitrite reductase, nirS, quite close to each other. In addition, the larger Ps. stutzeri cluster contains the nitrous oxide reductase gene, nosZ, and associated genes nosDFY, which are thought to be involved in biosynthesis of the enzyme. The nosZ gene has also been sequenced in P. denitrificans and appears to be associated with other nos genes as in Ps. stutzeri.
Each organism contains a set of homologous genes that tend to be associated with the structural gene for a denitrification enzyme and appear to be involved with its biosynthesis. In P. denitrificans, the genes downstream of nirS, nirECFD, have all been inactivated by insertional mutagenesis and this results in a cytochrome cd\(_1\) that lacks d\(_1\) haem; hence these genes are assigned a role in haem d\(_1\) biosynthesis [141]. Genes of this type that are conserved between the three clusters include nirF (a putative “scaffold” protein for haem d\(_1\) assembly), nirC (a small c-type cytochrome) and nirE (a S-adenosylmethionine uroporphyrinogen (III) C-methyl transferase).
A third class of genes encodes putative electron transport proteins and these tend to be less highly conserved between species. Examples include nirM, which encodes the Class I cytochrome c\(_{551}\) in Ps. aeruginosa [170] and nirT, encoding a membrane-bound tetrahaem cytochrome c in Ps. stutzeri [138]. The latter protein is not found in the other denitrification clusters, but has a homologue, napC, in the periplasmic nitrate reductase operon of T. pantotropha.
Each cluster contains several genes that appear to be involved in the regulation of denitrification. Predominant amongst these are members of the FNR family of proteins, discussed more fully in Section 1.7.1. Binding sites for these proteins are also detectable upstream of several denitrification operons. In addition, two other regulatory proteins are of note. One of these is nirQ [171] (designated norQ in P. denitrificans [172]). Inactivation of nirQ abolishes nitrite and nitric oxide reduction in vivo though the effects are complex; in Ps. stutzeri nitric oxide reductase becomes expressed to a two-fold higher degree and nitrite reductase retains in vitro activity. NirQ has some sequence similarity to NtrC, part of a two-component regulatory system which is linked to RNA polymerase containing the alternative sigma factor \(\sigma^{54}\). The other protein is nosR [173] (with a second homologue, nirI in P. denitrificans, de Boer et al. 1994). This is predicted to be a membrane-bound protein and may possess an iron-sulphur cluster. Inactivation of nosR in Ps. stutzeri results in the loss of nitrous oxide reductase and in a P. denitrificans nirI mutant, the nirS promoter is inactivated and cytochrome cd\(_1\) is absent (de Boer, 1996).
Finally, there are a number of genes to which no function can be assigned, or that do not appear to be involved in denitrification. One such example is nirJ from P. denitrificans, insertional mutagenesis of which has no effect on anaerobic growth [141]. Other examples include some recently sequenced open reading frames in Ps. stutzeri, such as orf247 (a short-chain alcohol dehydrogenase) and orf57, which has homology to a hypothetical E. coli protein, YEBC [174].
The main feature of the denitrification gene clusters identified to date is that they contain homologous genes, but the gene order and content of the clusters are not conserved. For instance the nor genes are upstream of the nir operon in Ps. aeruginosa and P. denitrificans, but are downstream in Ps. stutzeri. Similar genes are also often transcribed in different directions amongst the three clusters. This has led to the description of a “mosaic” gene organisation in these clusters [168] and supports a recent proposal that gene order is not conserved in evolution [175]. A final feature of note is that genes for the membrane-bound and periplasmic nitrate reductases, nar and nap respectively, are not found within the large denitrification gene clusters described above.
1.7 Regulation of denitrification
Respiratory N-oxide reduction is an anaerobic process and its regulation might therefore be expected to be under the control of both oxygen tension and ambient N-oxide concentration. Both of these aspects have been investigated, particularly in E. coli due to its genetic amenability, though similar systems are becoming apparent in the denitrifying bacteria. It is now clear that the control systems involved act largely at the level of transcription. Several systems comprising DNA-binding transcriptional activators (or repressors), and the sequence motifs to which they bind, are now understood in some detail and are described in the following sections.
1.7.1 Anaerobic response regulators: the FNR family
The anaerobic regulator FNR was first identified in mutants of E. coli, which were deficient in anaerobic growth on fumarate and nitrite [176]. Complementation analysis of these mutants revealed an open reading frame encoding the FNR protein. The protein has a subunit mass of around 28 kDa and shows high homology to the cyclic AMP receptor protein [177]. The most highly conserved regions are a DNA-binding helix-turn-helix motif, an N-terminal \(\beta\)-roll and an RNA polymerase contact site. However, the FNR protein contains an N-terminal extension with four cysteine residues, and a further internal cysteine at position 122. The function of the cysteines is to bind iron; the activity of FNR can be correlated to its iron content, which varies with the method of purification, and can be reconstituted to some degree [178]. More recently, it has been suggested that the N-terminal region forms an iron-sulphur cluster, which is labile in the presence of oxygen [179].
FNR is a transcriptional regulator which can act under anaerobic conditions in either a positive or a negative fashion, depending on the target gene. It has been suggested that FNR senses oxygen directly, possibly through a different binding affinity for Fe(II) and Fe(III) [180], but FNR-dependent promoter activity can also be stimulated by varying the redox potential in the cell [181], suggesting it may react more indirectly to the redox state. However, currently the consensus of opinion supports the idea that the inactive form of FNR is generated as a result of the destruction of its iron-sulphur cluster in the presence of oxygen [179]. Under anaerobic conditions, FNR is believed to dimerise and bind to a target sequence upstream of the FNR-dependent gene. Many such gene clusters have been identified in E. coli and other bacteria; the consensus target sequence (or “FNR box”) is an inverted repeat, TTGATN\(_4\)ATCAA and in E. coli, this is generally centred at about -41.5 nucleotides relative to the transcription start point [182] in genes that are activated by FNR. DNA-binding by FNR can then enhance transcription from the downstream promoter by contacting RNA polymerase. FNR can also negatively regulate transcription by binding at a different position and blocking RNA polymerase. The fnr gene is itself negatively auto-regulated in this way [183]. Binding to the FNR box has been demonstrated by a number of methods, including DNase I footprinting [184] and electrophoretic mobility shift assays [185]. Having identified FNR-dependent promoters, reporter gene fusions have been employed to measure induction or repression at the promoter under different growth conditions [186].
Site-directed mutagenesis has been used extensively to determine both the key amino acids in the FNR protein and the nucleotides at its binding site. This work has identified the conserved regions in the N-terminal cysteines of FNR, the RNA polymerase contact site and the DNA-binding domain. Mutations in residues 81-87 of the E. coli FNR protein can give a protein which is competent for DNA binding but not transcriptional activation, establishing the concept of a positive control “patch” which interacts with RNA polymerase [187]. In this region, Gly-85 and Gln-88 are invariant but residues 86 and 87 vary in other members of the FNR family (see below). Site-directed mutagenesis has also established the residues thought to be critical in the DNA-binding domain; this is the most highly conserved region of the FNR family [188]. A number of FNR homologues have now been identified that fall into several distinct groups (see below) but intriguingly, the consensus FNR box appears to be very similar regardless of which FNR homologue it binds. There may be some promiscuous binding between FNR proteins and their target sequences, but on the whole the homologues do not appear to substitute for one another in vivo, suggesting rather subtle differences in the specificity of the different FNR-like proteins. However, only two amino acid substitutions in FNR allows binding to a CRP consensus site (and vice-versa, [189]), and only three base changes in the FNR binding site allow binding by the CRP protein [190].
Much of the detailed biochemical and genetic characterisation of FNR has been carried out in E. coli, the organism in which it was first identified. More recently, a number of FNR homologues have been identified in several other species of bacteria and the FNR binding consensus has been found upstream of a large number of open reading frames. Many of these FNR homologues have been cloned and sequenced. Phylogenetic analysis of the sequences has revealed a superfamily of proteins (Figure 1.7).
![Unrooted phylogenetic tree of the FNR family of transcriptional activator proteins. Protein sequences that showed a high similarity to the FNR protein from _E. coli_ were retrieved from the GenBank database and aligned using the program Clustal W (Thompson _et al_. 1994 [@thompson_clustal_1994]). The phylogenetic tree was constructed by the method of neighbour-joining (Saitou and Nei, 1987 [@saitou_neighbor-joining_1987]) and analysed by bootstrapping (Felsenstein, 1985 [@felsenstein_confidence_1985]) using 1000 trials. The final tree was displayed using the program Treeview (Page, 1996 [@page_treeview_1996]). More details of these methods are given in Chapter 3. The tree shows that the FNR family falls into three main groups (van Spanning _et al_., 1997 [@van_spanning_fnrp_1997]): (A) the FNR subgroup, (B) the FixK subgroup and (C) the NNR subgroup. The FixK subgroup can be subdivided further into two groups, depending on the presence of an N-terminal cysteine cluster (van Spanning _et al_., 1997 [@van_spanning_fnrp_1997]). The NtcA proteins (Frías _et al_. 1993 [@frias_general_1993]) also cluster together within the NNR subgroup. Abbreviations: Syn1, _Synechococcus_ sp. WH7803, Psa, _Pseudomonas aeruginosa_, Ec, _Escherichia coli_, Sp, _Shewanella putrefaciens_, Bp, _Bordetella pertussis_, Av, _Azotobacter vinelandii_, Rh, _Rhizobium_ sp. IC3342, Ap, _Actinobacillus pleuropneumoniae_, Bj, _Bradyrhizobium japonicum_, Rp, _Rhodopseudomonas palustris_, Pd, _Paracoccus denitrificans_, St, _Salmonella typhimurium_, Rl, _Rhizobium leguminosarum_, Hi, _Haemophilus influenzae_, Rs, _Rhodobacter sphaeroides_, Tf, _Thiobacillus ferrooxidans_, Re, _Rhizobium etli_, Cy, _Cyanothece_ sp. ATCC 51142, Ac, _Azorhizobium caulinodans_, Syn2, _Synechococcus_ sp. PCC 7942, Syn3, _Synechocystis_ sp. PCC 6803, An, _Anabaena_ sp. PCC 7120, Rm, _Rhizobium meliloti_, Pss, _Pseudomonas stutzeri_ Zobell, Bs, _Bacillus subtilis_.](figure/fig1-7.png)
Figure 1.7: Unrooted phylogenetic tree of the FNR family of transcriptional activator proteins. Protein sequences that showed a high similarity to the FNR protein from E. coli were retrieved from the GenBank database and aligned using the program Clustal W (Thompson et al. 1994 [191]). The phylogenetic tree was constructed by the method of neighbour-joining (Saitou and Nei, 1987 [192]) and analysed by bootstrapping (Felsenstein, 1985 [193]) using 1000 trials. The final tree was displayed using the program Treeview (Page, 1996 [194]). More details of these methods are given in Chapter 3. The tree shows that the FNR family falls into three main groups (van Spanning et al., 1997 [195]): (A) the FNR subgroup, (B) the FixK subgroup and (C) the NNR subgroup. The FixK subgroup can be subdivided further into two groups, depending on the presence of an N-terminal cysteine cluster (van Spanning et al., 1997 [195]). The NtcA proteins (Frías et al. 1993 [196]) also cluster together within the NNR subgroup. Abbreviations: Syn1, Synechococcus sp. WH7803, Psa, Pseudomonas aeruginosa, Ec, Escherichia coli, Sp, Shewanella putrefaciens, Bp, Bordetella pertussis, Av, Azotobacter vinelandii, Rh, Rhizobium sp. IC3342, Ap, Actinobacillus pleuropneumoniae, Bj, Bradyrhizobium japonicum, Rp, Rhodopseudomonas palustris, Pd, Paracoccus denitrificans, St, Salmonella typhimurium, Rl, Rhizobium leguminosarum, Hi, Haemophilus influenzae, Rs, Rhodobacter sphaeroides, Tf, Thiobacillus ferrooxidans, Re, Rhizobium etli, Cy, Cyanothece sp. ATCC 51142, Ac, Azorhizobium caulinodans, Syn2, Synechococcus sp. PCC 7942, Syn3, Synechocystis sp. PCC 6803, An, Anabaena sp. PCC 7120, Rm, Rhizobium meliloti, Pss, Pseudomonas stutzeri Zobell, Bs, Bacillus subtilis.
Examples include NNR from P. denitrificans [197], ANR and DNR from Ps. aeruginosa [198,199] and FixK from Rhizobium meliloti [200]. FNR homologues have also been identified in Gram-positive bacteria [201] and the FNR box has been identified upstream of the CYP 55 gene in the fungus F. oxysporum [202].
Figure 1.7 shows that the FNR family falls into three main groups, which van Spanning et al. [195] have labelled types A, B and C, with a further subdivision of group B into B1 and B2. This grouping reflects the different functional domains within the proteins. Group A are the prototype FNR proteins containing the N-terminal cysteine cluster, RNA polymerase contact site, dimerisation domain and DNA-binding helix-turn-helix motif. Group B1 also contains all of these domains, but in groups B2 and C (the FixK and NNR subfamilies), the cysteine cluster is not present and there are differences in the putative RNA polymerase contact site. The lack of an iron-binding cysteine cluster is intriguing, as it is this that is proposed to respond to anaerobiosis in the FNR proteins. FixK and NNR clearly do respond to anaerobiosis in some way, as shown by strains carrying mutations in these genes [203,204], but the mechanism of the response is at present not clear. The differences in the RNA polymerase contact site have led some groups to suggest that the promoter regions of genes controlled in this manner bind an alternative sigma factor on RNA polymerase [205]. This is discussed further in Chapter 7. In many cases different FNR homologues have been found in the same organism; for instance in P. denitrificans, FnrP is thought to regulate transcription of membrane-bound nitrate reductase, cytochrome c peroxidase and cbb\(_3\) cytochrome oxidase, whilst NNR controls the nitrite and nitric oxide reductases [195]. Similarly in Ps. aeruginosa, the homologues ANR and DNR both seem to control denitrification genes [199], whilst in Ps. stutzeri, mutation of FnrA (an ANR homologue) has no effect on denitrifying growth, suggesting the presence of a second FNR-like protein [206].
1.7.2 The ArcA/B system
In E. coli a second system for oxygen-regulated transcriptional control is known, the ArcA/B system. This is a two-component sensor/regulator signal transduction system, of a type commonly found where response to an environmental signal is required [207]. ArcB is a transmembrane protein, with a periplasmic N-terminal sensor domain and a cytoplasmic C-terminal transmitter domain [208]. ArcA is a cytoplasmic protein, with an N-terminal receiver domain and a C-terminal DNA-binding domain [209].
The action of the ArcA/B system has been recently reviewed in Unden et al. [210]. Under low oxygen tension, the transmembrane protein ArcB responds indirectly to a signal at the cytoplasmic membrane (possibly the UQH\(_2\)/UQ ratio) and is autophosphorylated at His-292 by ATP. The phosphate is transferred intramolecularly to Asp-576, followed by a second phosphorylation at His-292. This double-phosphorylated species is then competent to phosphorylate the cytoplasmic ArcA protein at an N-terminal Asp residue. Once phosphorylated, ArcA becomes active and can bind DNA upstream of the genes to be regulated. ArcA-P acts for most genes as a repressor, blocking transcription under anaerobic conditions. Genes affected in this way include those encoding the citric acid cycle enzymes. ArcA-P can also act as an activator; the E. coli cydAB operon encoding cytochrome d oxidase possesses two ArcA-P binding sites upstream of its promoter, which have been identified by DNase I footprinting [211]. This work has lead to the identification of a consensus ArcA-P binding site. LacZ reporter gene fusions have been used to show that the Arc A/B system exerts its effects in the range 10-20% air saturation, whereas Fnr functions in the 0-10% range [212]. Hence in E. coli the two systems co-ordinate the control of respiratory gene expression to ensure optimal use of oxygen as environmental conditions change.
Unlike FNR the ArcA/B system is only well characterised in E. coli and to date, none of the denitrification genes have been shown to be under ArcA/B control. However, the system is important as a complementary response to the FNR system and it is possible that something similar will be found in denitrifying bacteria.
1.7.3 Sensors of nitrate and nitrite
In E. coli nitrate and nitrite are sensed by two, interacting, two-component regulatory systems known as NarX/L and NarQ/P [213,214]. These function in a similar fashion to the ArcA/B system described earlier. NarX and NarQ are transmembrane proteins, possessing a periplasmic domain which can sense nitrate or nitrite. A region within this domain termed the “P-box” has been shown by site-directed mutagenesis to be involved in nitrate/nitrite sensing [215]. This results in an autophosphorylation at the C-terminal cytoplasmic domain, which in turn goes on to phosphorylate the cytoplasmic component, NarL or NarP. Activated NarL/P can then go on to bind to a target DNA sequence and enhance or repress transcription of nitrate/nitrite dependent genes. A consensus site, the “NarL heptamer”, has been proposed with the recognition sequence TACYNKT (where Y=C or T, N=any nucleotide and K=A or C) [216]. The NarL heptamer appears as a single heptamer, a direct or inverted repeat, in either orientation and apparently between +20 and -220 nucleotides from the transcription start site. The interaction between the homologous NarX/L and NarQ/P systems is quite complex; essentially it seems that nitrate stimulates NarX/Q to phosphorylate NarL/P, nitrite stimulates NarQ to phosphorylate both NarL and NarP and finally that NarX can act to dephosphorylate NarL-phosphate in the absence of nitrate [214].
No analogous system has yet been identified in the denitrifying bacteria, but all four N oxides have been reported to increase the expression of at least one reductase, particularly in Ps. stutzeri and Ps. aeruginosa. In the latter organism, expression of the cytochrome cd\(_1\) nitrite reductase appears to rely more on the presence of nitrate or nitrite than on anaerobiosis [217]. Levels of nitrous oxide reductase in Ps. stutzeri are also regulated by nitrate, nitrous oxide and anaerobiosis [218]. There also appears to be a regulatory link between the nitrite and nitric oxide reductases. In Ps. stutzeri, mutation in nitric oxide reductase reduces nitrite reductase activity, though its expression level is similar. The converse is true for mutants in nitrite reductase [76]. This implies that nitric oxide may induce its reductase and that there is a functional coupling between these two enzymes.
It seems likely that control of denitrification will prove to be a complex process, with several hierarchical levels that act to “fine-tune” the response to changing environmental conditions. This is already becoming apparent in the systems described previously for E. coli. One example of a complex promoter system is the focApfl operon, encoding pyruvate formate lyase and the focA gene product. This system is regulated by both ArcA and FNR, which can act at several different promoters in the operon. One promoter, P6A, overlaps an FNR-dependent promoter, modulating its activity by competition for FNR- and RNA polymerase-binding [219]. There are also several examples of promoters that are regulated by the FNR, Arc A/B and NarX/L components [220].
Although the regulation of denitrification is known to occur primarily at the level of transcription, transcriptional analysis of denitrification genes has been reported in relatively few cases. Northern blot analysis has been reported for the norBC genes of Ps. stutzeri which are co-transcribed [76], and for the nosZ gene which is apparently under the control of 6 alternate promoters [221]. Also in Ps. stutzeri, transcription of the nirS gene for cytochrome cd\(_1\) nitrite reductase occurs as two transcripts, nirS and nirST, and is controlled by anaerobiosis and nitrate [222]. The fnrA gene from this organism is also known to be transcribed as a single 0.85 kb species [76]. Induction of the mRNAs for nitrate, nitrite and nitrous oxide reductases in P. denitrificans during the switch to anaerobiosis has also been reported [56]. More commonly, transcriptional and translational features are inferred from inspection of the gene sequence; hence in P. denitrificans, the nirE and nirC genes have overlapping stop and start codons [141], implying that they are co-translated, whilst stem-loop analysis of the nir operon in Ps. aeruginosa has been used to suggest that the 11 genes nirSMCFDLGHJEN are transcribed on a polycistronic mRNA [223].
1.8 Aims of the present work
At the outset of this project the X-ray crystal structure of cytochrome cd\(_1\) nitrite reductase from T. pantotropha had been determined from diffraction data and the P. denitrificans PD1222 sequence, with no complete protein sequence from the T. pantotropha protein. The initial aim of the project was to clone the structural gene, nirS, from T. pantotropha, so as to obtain the polypeptide sequence and refine this crystal structure. Mapping and sequencing of the nirS gene and surrounding regions was also essential for subsequent DNA manipulations. It was also of interest to determine whether the organisation of the nir cluster is similar to that of the closely-related P. denitrificans.
Having cloned the nirS gene, the next aim would be to investigate several expression systems for the enzyme. This work would involve cloning the gene into a suitableexpression vector and trying to transform a number of heterologous hosts, to determine which ones expressed the protein most efficiently. As the d\(_1\) haem group is unique to cytochrome cd\(_1\), it would also be advantageous to use heterologous hosts which are able to synthesise this group, so as to express the active holoprotein. Development of a suitable expression system is an essential prerequisite for future site-directed mutagenesis studies.
Finally, transcriptional regulation of the nirS and associated genes is of interest. Knowledge of this process may help to understand the expression of the protein, so a third aim of the project was to investigate the mRNA transcribed from the nirS gene, using methods such as Northern blotting and determination of the transcription start site. This work was expected to help to define novel promoter regions in the denitrification genes of P. denitrificans and T. pantotropha, and to provide insight into the transcriptional regulation of denitrification.