A Comparison of the \(\beta\)-propeller structure in cytochrome cd\(_1\) with other \(\beta\)-propeller structures
Residues 135-567 of T. pantotropha cytochrome cd\(_1\) form a rigid \(\beta\)-propeller domain which provides a binding pocket for the d\(_1\) haem. The propeller structure consists of an eightfold repeat of a four-stranded antiparallel \(\beta\)-sheet motif and is so named because each set of four strands is arranged in a circular domain, resembling a propeller with eight blades. A schematic diagram of this domain is shown in Figure A1.
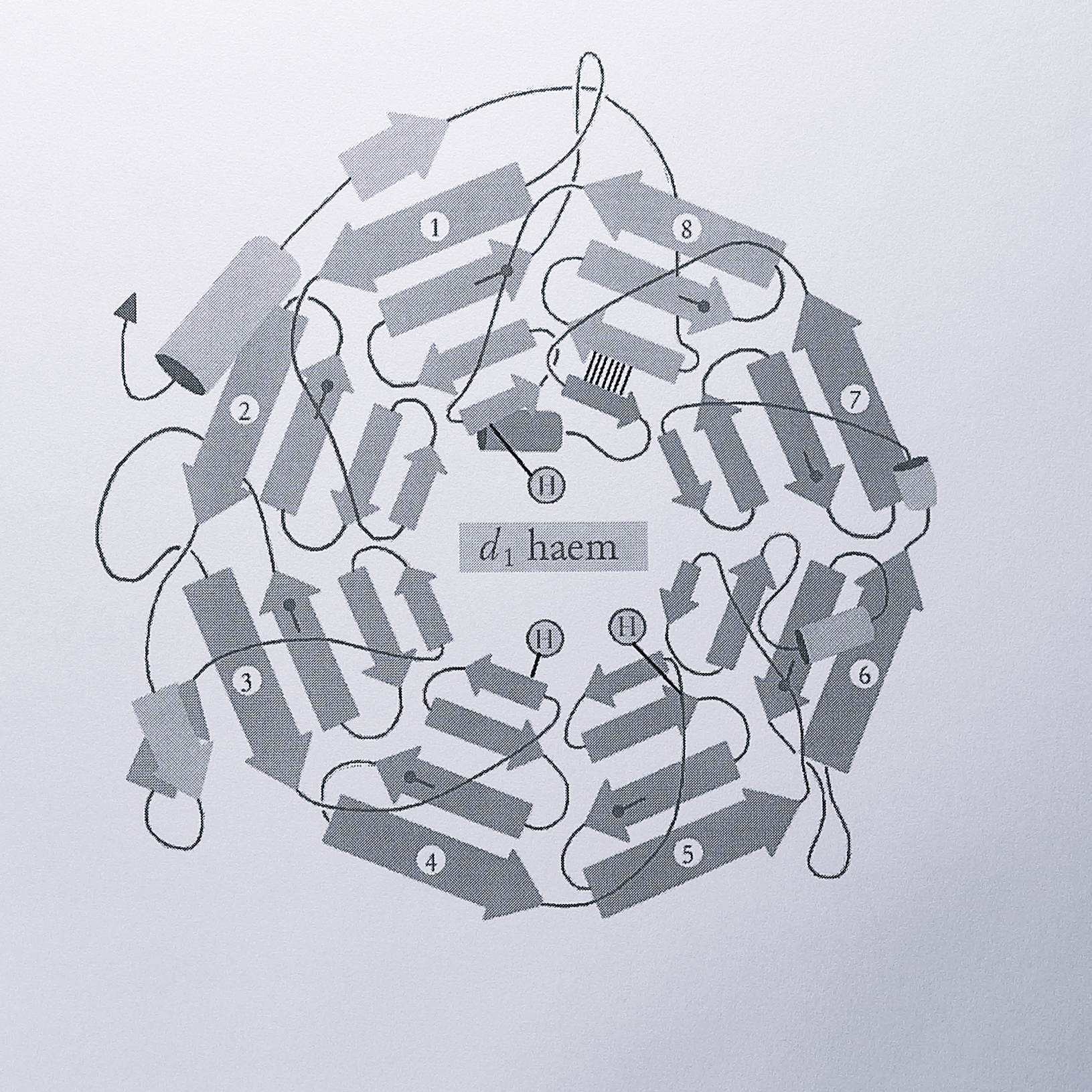
Figure A.1: Schematic diagram of the \(\beta\)-propeller d\(_1\)-haem binding domain of T. pantotropha cytochrome cd\(_1\). The symbol H represents histidine residues; on blade 1, His-200, blade 4, His-345 and blade 5, His-388. The “Velcro” closure of the ring is indicated by six parallel lines between the 2 \(\beta\)-strands of blade 8, each line representing a hydrogen bond. The lollipop symbol represents conserved aspartate residues at the end of strand 3 within each blade.
The sheets are packed face to face, providing structural stability via mainly hydrophobic interaction between them. The rigidity of the propeller structure is also maintained by interactions that act to close (or “Velcro”) the propeller ring. In cytochrome cd\(_1\) this is achieved by forming the last blade (conventionally assigned as the blade where closure occurs) from both termini of the propeller domain; the C-terminal innermost strand of blade 8 bonds via six main chain hydrogen bonds to one of the other three antiparallel sheets which are provided by the N-terminus.
Propeller structures are being found in an increasing number of proteins, from both prokaryotes and eukaryotes. Cytochrome cd\(_1\) provides only the second example of an eight bladed propeller, the other being that of the heavy subunit of methanol dehydrogenase, the structure of which has been determined from Methylobacterium extorquens [355] and Methylophilus methylotrophus W3A1 [356]. Other propeller structures are known with seven blades (fungal galactose oxidase from Dactylium dendroides [357], methylamine dehydrogenase from P. denitrificans [153], and eukaryotic G-protein \(\beta\)-subunit, [358]), six blades (influenza neuraminidase [154] and bacterial sialidase [359]) and four blades (hemopexin [360] and collagenase [361]). All propeller domains contain a central tunnel, preserved by the rigidity of the structure, which is used toco-ordinate a ligand or prosthetic group involved in catalysis or some other protein function. In the case of cytochrome cd\(_1\), the channel is filled by the d\(_1\) haem, which makes a large number of contacts with protein side chains and solvating water molecules lining the channel.
The emergence of several \(\beta\)-propeller structures prompted us to ask whether the cytochrome cd\(_1\) crystal structure and sequence indicates any determinants that could account for such structures. This work was mostly carried out by Drs. Vilmos Fülöp and Simon Baker, but it results from analysis of the cytochrome cd\(_1\) sequence reported in Chapter 3 and is published in a joint paper [264], and so it is described here in some detail.
Direct sequence comparison of cytochrome cd\(_1\) to methanol dehydrogenase, the other example of an eight-bladed propeller protein, reveals no significant homology. Despite this, the d\(_1\) haem binding domain can be very closely superimposed onto the eight-bladed propeller of methanol dehydrogenase, using the program STAMP [362], an alignment program for 3-D structures, rather than primary sequences. Essentially the same propeller can therefore be achieved with two different sequences, though in MDH ring closure is achieved differently; the last blade is formed from three C-terminal antiparallel sheets and an outermost sheet from the N-terminus. This feature is also seen in the other seven and six-bladed propeller structures. Visual inspection of the cytochrome cd\(_1\) protein sequence reveals that the third sheet of each blade almost always ends with an aspartate residue (residues 183, 224, 272, 326, 370, 474 and 535, with Asp 420 displaced two residues from the C-terminal end of blade 5, sheet 3), as shown in Figure 3.13. These residues are predominantly exposed to the solvent and seven of the eight are superimposable on each other when the third sheets from each blade are aligned. Methanol dehydrogenase (MDH) is one of a family of bacterial dehydrogenases termed the PQQ-binding proteins (after the pyrolloquinoline quinone cofactor that they possess), which includes low specificity alcohol dehydrogenases (ADH) and periplasmic glucose dehydrogenases (GDH). Alignment of 15 sequences from this family to M. extorquens MDH gives 23-98% identity, suggesting a common fold in the PQQ-binding domain; it has been suggested previously [363] that the \(\alpha\)-subunit of ADH from Acetobacter aceti will possess an MDH-like eight-bladed \(\beta\)-propeller domain.
The STAMP program was used to overlay the structural primary sequence alignment of the MDH and cytochrome cd\(_1\) \(\beta\)-propeller domains onto the 15 PQQ-binding protein sequences. This allowed the prediction of the positions of seven of the eight blades in these sequences (the eighth is harder to predict due to the differing method of ring closure described previously). The alignment revealed terminal \(\beta\)-sheet aspartate residues in four out of eight blades in the MDH sequences, at identical positions to those in cytochrome cd\(_1\). Of the 105 predicted propeller blades (seven blades in the 15 sequences), 60 contained a terminal \(\beta\)-sheet aspartate residue, preceded by hydrophobic amino acids. In contrast, 37 of the predicted blades contained a previously identified feature named the “tryptophan docking motif” [355]. This feature was first noted in methanol dehydrogenase and consists of a stabilising stacking interaction between the indole ring of a tryptophan residue on one propeller blade and a glycine residue on an adjacent blade. The feature is entirely absent from cytochrome cd\(_1\) and cannot therefore be essential for propeller stability. The weak consensus derived from common residues in all the blades of the d\(_1\) haem domain was also used to search for possible propeller structures in the protein sequence databases with less success. It is concluded that the terminal aspartate motif plays a common role in cytochrome cd\(_1\) and the PQQ-binding proteins, but at present this role is unclear. The aspartates are exposed to the solvent and a role in structural integrity seems unlikely; they may be involved in the pathway of folding in the periplasm.
Although there seem to be relationships between the PQQ-binding proteins and the 8 bladed propeller of cytochrome cd\(_1\), these cannot be related to a number of other features in known propeller structures. In addition to the aforementioned tryptophan docking motif, tryptophan residues are also involved in a large family of proteins called the “WD repeat” family, which includes the \(\beta\)-subunit of the eukaryotic G-proteins [364]. In these propeller structures, long repeats are found that terminate with the residues Trp-Asp, at the end of the third strand in each blade. The Trp residue is buried between sheets and interacts with an Asp-His-Ser/Thr triad, whilst the Asp residue stabilises the loop between the third and fourth strands of a blade by a number of hydrogen bonds. Again, this is very different to any structural motif found in cytochrome cd\(_1\) or other propeller proteins, although Sondek et al. (1996) [358] have noted a weak sequence similarity between the WD-repeat proteins and some of the more strongly conserved repeats in fungal galactose oxidase. However, the third strand of each blade does terminate with Asp in the WD repeat proteins.
The six-bladed propeller protein bacterial sialidase contains a consensus repeat sequence Ser/Thr-Xaa-Asp-[Xaa]\(_n\)-Gly-Xaa-Thr-Trp/Phe, termed the “Asp box” [359]. This appears to dictate the fold and integrity of the propeller and again, the Asp residue falls near the end of strand 3 in each \(\beta\)-sheet. However, the Asp box is not present in cytochrome cd\(_1\). A final type of motif, known as Drosophila kelch, has been identified in mouse MIPP protein, poxvirus and fungal galactose oxidase [365], where one motif is found in each of the seven blades. This does not contain the Asp residue prevalent in the other motifs and cannot be used to describe cytochrome cd\(_1\). Finally, the four-bladed propeller structures (hemopexin and collagenase) are quite differently arranged to the other examples, in that adjacent strands in a sheet are perpendicular and not stacked on one another. Stabilisation in these structures is brought about by a calcium ion and a disulphide bond between the first and last blades. These features are not relevant to cytochrome cd\(_1\).
In summary, although the propeller domain is a common feature to several proteins, the fold seems to be achieved in a number of different ways which are not dictated by sequence features common to all the proteins. Aspartate residues in cytochrome cd\(_1\) and a number of other proteins, particularly the PQQ-binding proteins, seem to be important in some as yet unknown way, but are not necessarily involved with the integrity of folding and do not form a clear basis for prediction of the fold from protein sequence alone. Each group of propeller proteins (cytochrome cd\(_1\) type, PQQ-type, WD repeat-type, Asp-box-type and kelch-type) possesses some conserved residues. However, given the wide distribution of propeller proteins, with no fundamental common sequence features even at a low level, the model of propeller formation by divergent evolution proposed by Bork and Doolittle (1994) [365] seems less likely. Murzin (1992) [366] suggests that propeller formation is merely a variation on the packing of \(\beta\)-sheets which can be extended from a consideration of the geometry that defines an ordinary \(\beta\)-sandwich protein and does not require any special sequence constraints. The analysis presented here seems to support this idea.