Features of the DNA sequence
The nirS gene and its immediate flanking regions are very similar in both T. pantotropha and the two previously published P. denitrificans sequences; in particular the nirS open reading frame from T. pantotropha is 94.1% identical to that of P. denitrificans PD1222. The GC content of the two genes is 64%, a typical figure for genes from Paracoccus species, reflecting the high GC content of the genome (68% [259]).
In P. denitrificans PD1222, an open reading frame was identified both upstream and downstream of the nirS gene. The upstream ORF was designated nirI and has some sequence homology to the regulatory protein nosR [96,141], in that it is predicted to be an integral membrane protein with a putative iron-sulphur cluster. It is transcribed divergently from the nirS gene. Insertional mutation in nirI results in the loss of cytochrome cd\(_1\) [141], Chapter 7. The nirI ORF is also present in the sequence from P. denitrificans IFO 12442, although Ohshima et al. (1993) [140] failed to identify it as such. Translation of the first 37 codons from the latter sequence shows 97% identity to the P. denitrificans PD1222 NirI protein, with a substitution of Phe for Leu at position 33. In the T. pantotropha sequence, the start of the nirI gene can also be discerned, with a substitution of Arg for Gly at position 4.
Downstream of nirs, de Boer et al. (1994) [141] identified the gene nirE. The product of this gene shows similarity to a number of S-adenosyl-L-methionine uroporphyrinogen (III) methyltransferases such as the sirohaem biosynthetic gene cysG of E. coli [42], and cobA, a cobalamin biosynthesis gene of S. typhimurium [260], and is thought to be involved in methyl transfer to the isobacteriochlorin ring during the biosynthesis of haem d\(_1\). Insertional inactivation of nirE results in the formation of inactive cytochrome cd\(_1\), lacking the d\(_1\) haem [141]. This open reading frame was identified (but not characterised) in P. denitrificans IFO 12442 [140].
Translation of the first 27 codons from the P. denitrificans IFO 12242 gene shows 81% identity to the P. denitrificans PD1222 NirE product. Once again, the start of the nirE gene can be identified in the T. pantotropha sequence. These sequence data indicate that the structure of the nir gene cluster in the immediate vicinity of nirS is highly conserved in T. pantotropha and the two strains of Paracoccus denitrificans characterised to date. Further mapping of this region, described in Chapter 4, confirms this observation.
The non-coding regions flanking the nirS gene contain a number of inverted repeats which are also quite highly conserved. In the T. pantotropha sequence, the first of these (repeat 1, Table 3.1) begins 81 bp upstream of the nirS initiation codon. In P. denitrificans PD1222 this sequence was identified as containing a putative binding site for NNR, a transcriptional activator protein belonging to the FNR family [188], Chapter 7. The NNR gene has been cloned and sequenced in this organism [197] and insertional mutagenesis results in the loss of nitrite and nitric oxide reductases. The putative NNR binding sequences are very similar, with a single substitution of T for A at position 12 in the T. pantotropha sequence. This lies in the non-conserved region of the NNR box consensus and is unlikely to be important. An identical sequence to the P. denitrificans PD1222 NNR box is found in the P. denitrificans IFO 12442 sequence, although Ohshima et al. (1993) [140] did not identify it as such. Instead, they assigned the TAA trinucleotide as a stop sequence for a putative upstream open reading frame and included part of the NNR box sequence in a larger, downstream repeat. Both of these assignments appear to be erroneous in the light of other data. Expression of cytochrome cd\(_1\) in all three organisms is, therefore, concluded to be transcriptionally regulated by the NNR protein. An alignment of these three NNR boxes with the FNR consensus sequence is shown in Figure 3.6.
Immediately downstream of the NNR box is a small, perfect inverted repeat (repeat 2, Table 3.2). This structure is also seen in the two P. denitrificans sequences but interestingly, in both cases, the first and last dinucleotides are substituted by CC and GG respectively. Hence the repeat structure is preserved, although the sequences differ. No putative function has been assigned to this repeat, but its structural conservation implies that it has some importance. A possible function is the binding of a transcriptional repressor protein; this possibility is explored more fully in Chapter 7.
Two more repeat sequences are found between the nirS and nirE genes in T. pantotropha. The first of these (repeat 3, Table 3.2) was identified in the P. denitrificans PD1222 sequence as a perfect 10 bp inverted repeat and is also found in the P. denitrificans IFO 12442 sequence. However, in T. pantotropha the sequence differs at positions five and seven and so is an imperfect repeat. The significance of this repeat is unclear.
Finally, a large imperfect repeat (repeat 4, Table 3.2) is found 6 bp downstream of repeat 3 in T. pantotropha. The sequence of this repeat is perfectly conserved in the P. denitrificans PD1222 sequence, but less so in P. denitrificans IFO 12442 where the corresponding sequence differs at positions one and five. In P. denitrificans PD1222 two assignments were made to this sequence; the first GGGGGC hexanucleotide was suggested to be a putative -10 promoter sequence for the nirE gene [141], based on the promoter consensus for purple non-sulphur bacteria of Steinrücke and Ludwig (1993) [259], whilst the structure as a whole was suggested by the same authors to be a putative oxygen responsive element (ORE). An ORE is a region of dyad symmetry found upstream of genes that are regulated by oxygen; it was first identified in photosynthetic genes from the genus Rhodobacter [261] and is thought to bind a protein that represses transcription under aerobic conditions. Steinrücke and Ludwig (1993) [259] have proposed that some Paracoccus are also ORE-regulated. Both of the assignments proposed by de Boer et al. (1994) [141] were tentative and both now appear unlikely. Based on the finding that nirS is transcribed as a single mRNA species (see Chapter 7 for data and discussion), this feature could be involved in transcriptional termination.
Relationship of the primary sequence to the structure of cytochrome cd\(_1\)
Sequencing of the nirS gene from T. pantotropha has allowed the crystal structure of the enzyme, originally deduced from electron density by comparison with the P. denitrificans PD1222 sequence [127], to be checked for errors and refined [264]. There were 14 discrepancies between the density map and the P. denitrificans PD1222 sequence in the original publication of the oxidised crystal structure; the translated protein sequence has confirmed 11 of these (three were shown to be the same as the P. denitrificans sequence), as well as revealing four others that were not apparent earlier (counting from the N-terminus of the mature protein these are K2OR, G101A, E339S and A499T, the corrected amino acid second). The oxidised crystal structure has now been determined at the higher resolution of 1.28 \(\unicode{x212b}\) and is fully consistent with the protein sequence presented in this chapter.
The crystal structure of cytochrome cd\(_1\) contains 559 residues, giving a calculated molecular weight of 62 249 Da per monomer including the covalently-bound c haem moiety. The monomeric molecular weight as measured by electrospray mass spectrometry was 63 091 +/- 8 Da [101], a discrepancy of 842 Da (when using the P. denitrificans sequence for comparison). From this, it was argued that eight N-terminal residues of the mature protein in the crystal structure were not visible, due to disorder in the crystal packing [127]. This has been confirmed by sequencing of the nirS gene. The T. pantotropha nirS gene encodes a protein of 596 amino acids, with a calculated molecular weight of 66 045 Da, including the c haem. The first 29 residues are predicted to be a cleavable periplasmic targeting sequence (Figure 3.3), giving a molecular weight for the mature protein of 63 141 Da. The discrepancy between the latter figure and the molecular weight calculated from the crystal structure is 892 Da, which is accounted for by the first eight residues in the mature T. pantotropha sequence. These residues are QEQVAPPK, an additional molecular weight of 896 Da. The N-terminus of cytochrome cd\(_1\) from T. pantotropha is known to be blocked, as it is resistant to Edman degradation [240]. An N-terminal glutamine, as postulated above, can account for this by cyclisation to pyroglutamate. The N-terminus of cytochrome cd\(_1\) from P. denitrificans IFO 12442 has also been reported to be blocked [140]. However, the equivalent eight residue sequence in the two Paracoccus sequences is QEQAAPPK and a further difference (counting from the N-terminus of the uncleaved protein, residue 27 is alanine in the Paracoccus sequences, valine in T. pantotropha) makes the predicted signal cleavage site one residue earlier in both Paracoccus sequences, giving AQEQAAPPK as the mature N-terminus. This would not be expected to show blockage, but in the absence of further experimental data, the predicted mature sequences have been used in this chapter and the effect on the alignment is insignificant.
The crystal structure of cytochrome cd\(_1\) from T. pantotropha showed that the c haem was ligated by His-17 and His-69 (numbering is with the targeting sequence removed) and the d\(_1\) haem by Tyr-25 and His-200 [127]. Both of these arrangements were somewhat unexpected; first, bis-histidyl ligation is normally associated with low-potential haems [151,265], whereas the c-haem in cytochrome cd\(_1\) has a midpoint redox potential of around 243 mV (A. Koppenhöfer, unpublished data). Second, tyrosyl ligation to haem groups, which is most common in bacterial catalases [266] also results in a low redox potential, as illustrated by mutant forms of haemoglobin [267] and myoglobin [268]. The midpoint redox potential of the d\(_1\) haem in cytochrome cd\(_1\) from T. pantotropha is not known but is likely to be a few tens of millivolts lower than that of the c haem (Chapter 1, Table 1.2). However, the protein sequence confirms the presence of all of these residues.
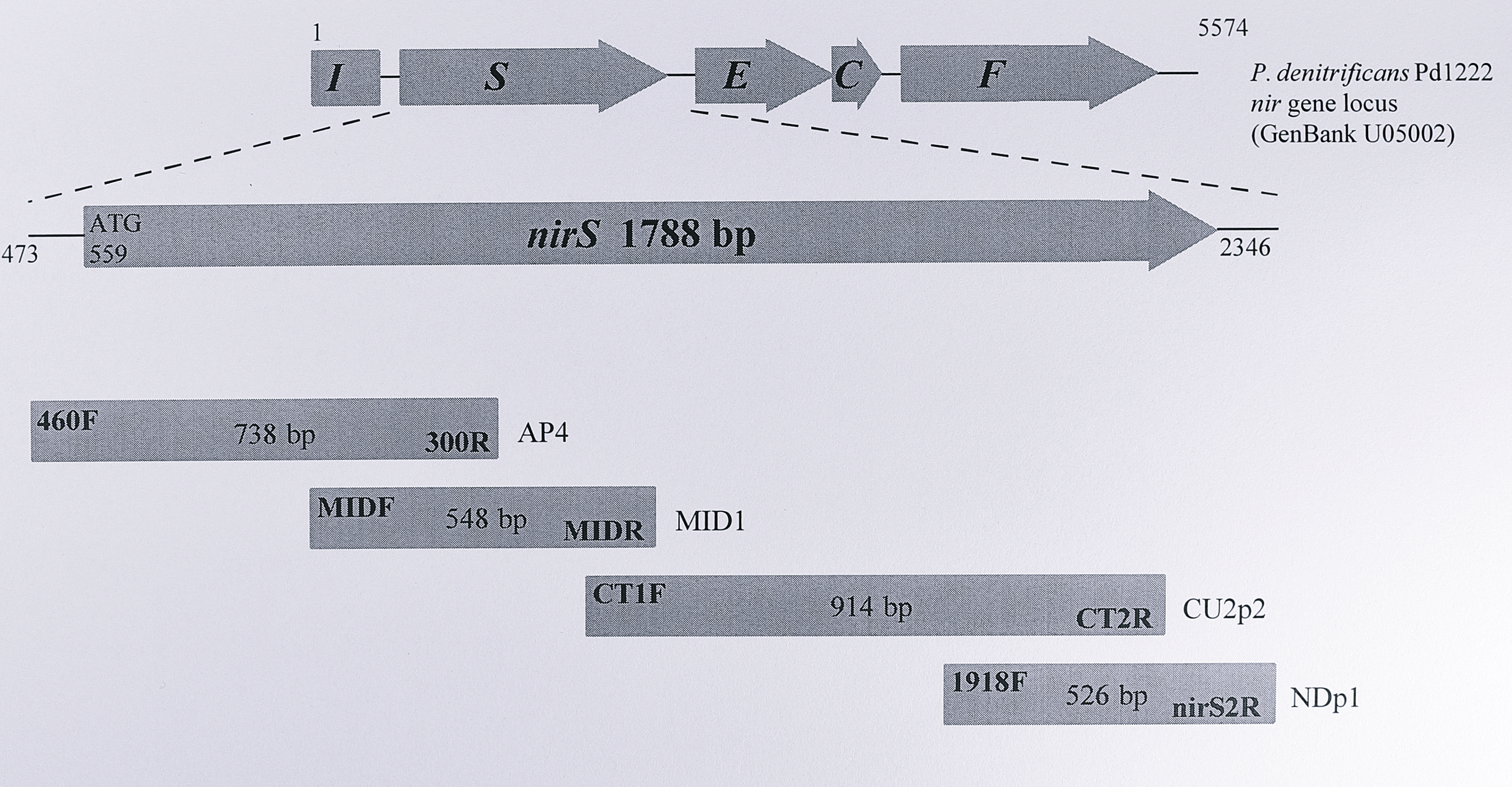
His-17 and Tyr-25 are located towards the N-terminus of the protein, which Figure 3.4 shows to be the least conserved region of the cytochromes cd\(_1\). Intriguingly, inspection of Figure 3.4 shows that these residues are conserved only in T. pantotropha and the two strains of Paracoccus. The other proteins lack the N-terminal extension seen in these sequences which contains the two haem ligands. Of the other proteins, only that from Ps. aeruginosa contains a histidine or tyrosine residue at even roughly similar positions in the N-terminus before the c haem binding site. This finding was extremely unexpected. Ligands to the haem groups control several important factors such as the redox potential, substrate accessibility and the reaction mechanism; in T. pantotropha, Tyr-25 has been postulated to play a crucial role in the release of nitric oxide from the haem group [127]. The importance of these residues and the overall high similarity of the proteins suggests that functional amino acids should be highly conserved. For this reason, it was decided to re-sequence the relevant segment of the nirS sequence from Ps. stutzeri Zobell so as to examine the possibility that the sequence had been mis-reported, or that errors in the sequence (such as base changes or frame shifts) could have resulted in the apparent absence of at least Tyr 25. Two sets of forward and reverse primers, flanking the N-terminal region of the nirS protein, were designed and used to amplify genomic DNA from Ps. stutzeri Zobell, as shown in Figure 3.7.
The four resulting PCR products were directly sequenced from low-melting point agarose using these primers. Figure 3.7 indicates the region, direction and strand coverage (forward, reverse or both) of the sequence obtained. This sequence was 100% identical to that deposited in the GenBank database. It is thus clear that the gene sequence is correct and that the tyrosyl and histidyl ligands in the protein from T. pantotropha really are absent from at least the Ps. stutzeri Zobell enzyme.
This result raises a number of possibilities. First, the c- and d\(_1\) haems in cytochrome cd\(_1\) in the oxidised state from Ps. stutzeri (and from other species) may be liganded by tyrosyl and histidyl residues lying elsewhere in the polypeptide chain. This would result in a substantially different tertiary structure in these enzymes and given the overall high sequence homology, does not seem to be a credible proposal. Second, the other cytochromes cd\(_1\) may have different haem ligands (such as the conserved methionines and histidines seen in Figure 3.4), which could preserve a similar structure to the T. pantotropha protein, but might be expected to result in different spectroscopic and biochemical properties. Third, the oxidised T. pantotropha enzyme structure may not be representative of the physiologically-active structure found in vivo; the latter, hypothetical, structure may have the haem ligands that are conserved between all cytochrome cd\(_1\) sequences.
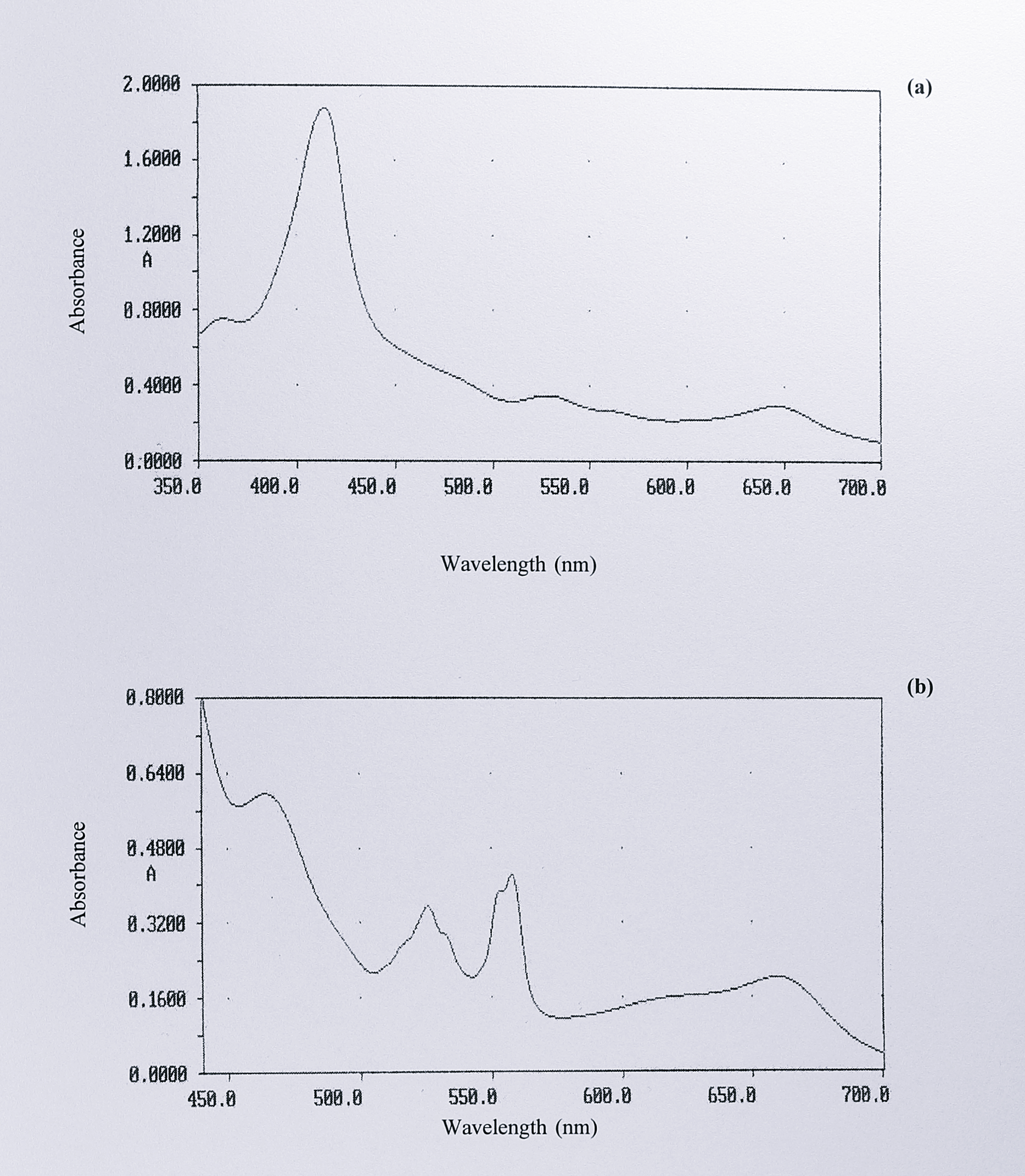
In order to examine the optical spectrum of cytochrome cd\(_1\) from Ps. stutzeri Zobell more closely, it was purified using the same procedure as that for the T. pantotropha protein. This protocol gave a product that was approximately 90-95% pure as judged by SDS-PAGE. The spectra of the oxidised and reduced forms of the enzyme are shown in Figure 3.8.
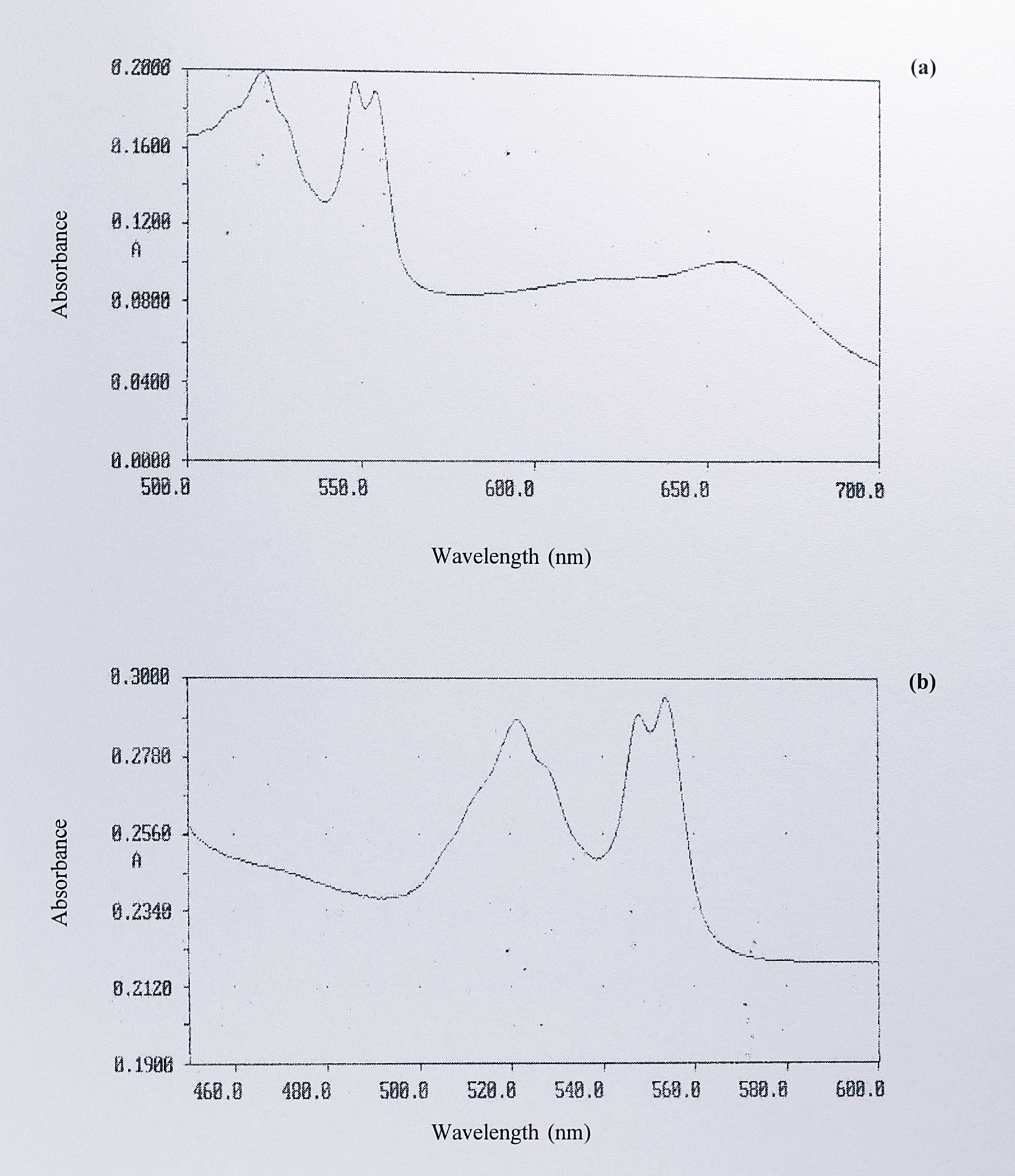
Overall, the spectra of the seven cytochromes cd\(_1\) that have been sequenced are very similar. In the oxidised form there are peaks at or near 411 and 640 nm, with shoulders around 561, 325 and 360 nm. In the reduced form, peaks at 418, 460, 521, 548, 554 and 625 nm (dithionite-reduced) or 655 nm (ascorbate-reduced) are seen. The absorbances at 655/625 and 460 nm are assigned to the d\(_1\) haem, whereas the 548-554, 521/525 and 418/411 nm peaks are the \(\alpha\), \(\beta\) and \(\gamma\) (Soret) peaks of the c-type haem. However, the enzyme from Ps. stutzeri Zobell shows a small, but interesting difference. In the split \(\alpha\)-peak of the reduced c haem at 548-554 nm, the first peak of the pair is slightly higher for the enzymes from T. pantotropha, P. denitrificans and Ps. aeruginosa. In the purified enzyme from Ps. stutzeri Zobell, this situation is reversed. This feature can be seen in published spectra by other workers [73] and was also specifically noted for the enzyme from Ps. stutzeri JM300 [253]. Interestingly, if the d\(_1\) haem is removed from the T. pantotropha protein to give the semi-apo enzyme, the relative heights of the split \(\alpha\)-peaks in the reduced state become reversed, so as to resemble the Ps. stutzeri enzymes (Figure 3.9).
From the T. pantotropha crystal structure, we know that the Tyr-25 ligand originates in the c haem domain. Hence this result could be interpreted as showing that the d\(_1\) haem domain has a subtle effect on the absorption spectrum of the c haem, which is altered when the d\(_1\) haem is removed. The apparent lack of this effect in the Ps. stutzeri enzymes might be taken as evidence that a similar link between the c and d\(_1\) haem domains does not exist in these proteins. A second interpretation is that d\(_1\) haem loss is more extensive during the purification of the Ps. stutzeri enzymes, yielding a species more like the semi-apo enzyme. However, the observation of this spectrum by other groups and previously reported activity measurements [253] argue against this idea.
In fact, mounting spectroscopic evidence indicates that haem ligation is genuinely not identical in different cytochromes cd\(_1\). Magnetic circular dichroism (MCD) and electron paramagnetic resonance (EPR) spectroscopy of the cytochrome cd\(_1\) from Ps. aeruginosa have indicated Met-His ligation at the c haem and His-His ligation at the d\(_1\) haem [124]. EPR and Mössbauer spectroscopy of the enzyme from Thiobacillus denitrificans suggested His-His ligation at both haems [126]. More recently, the T. pantotropha and Ps. stutzeri proteins in the oxidised form have been compared using EPR and MCD spectroscopy at the University of East Anglia [125]. The latter method in particular is regarded as the most accurate diagnostic of haem ligation [269]. The EPR and MCD data are interpreted as being consistent with His-His ligation at the c haem in the T. pantotropha enzyme although, interestingly, a very small proportion of the sample appears to have Met-His ligation. The d\(_1\) haem ligands are more difficult to assign in the absence of data from model compounds, but are thought to be consistent with His-Tyr or possibly His-OH ligation in both cases. However, the c haem in the oxidised Ps. stutzeri enzyme is clearly Met-His ligated, as was previously reported for cytochrome cd\(_1\) from Ps. aeruginosa [124].
In summary, it is apparent that there are significant differences in haem ligation amongst the cytochromes cd\(_1\), though bis-histidyl ligation at the d\(_1\) haem and methionyl histidyl ligation at the c haem are commonly assigned to several members of the group. This brings us to the question of whether the T. pantotropha cytochrome cd\(_1\) crystal structure is anomalous or in some way misleading. There can be no doubt that the structure is “correct” in terms of the integrity of the diffraction data, its interpretation, the chain tracing methods used and correlation with the protein sequence; the question is whether the structure is physiologically relevant.
As described above, MCD spectra of the T. pantotropha enzyme confirm the bis histidyl ligation seen in the oxidised crystal structure, whereas the enzymes from Ps. stutzeri and Ps. aeruginosa are thought to have Met-His ligation to the c haem in the oxidised state. Recent work [166] has shown that in reduced crystals of cytochrome cd\(_1\) from T. pantotropha (which do not crack when reduced by sodium dithionite, unlike crystals of the Ps. aeruginosa enzyme [147], dramatic changes in haem ligation are observed. Tyr-25 is seen to move away from the d\(_1\) haem, as predicted by spectroscopic and mechanistic studies (Chapter 1, Section 1.5.4), but at the c haem His-17 is no longer a ligand. Instead, a loop of residues (100-114) moves upwards in the structure and Met-106 becomes a new c haem ligand. This arrangement means that the T. pantotropha enzyme in the reduced state more closely resembles the predicted ligation of the enzymes from Ps. aeruginosa and Ps. stutzeri; additionally Met-106 is a conserved residue in all cytochrome cd\(_1\) sequences.
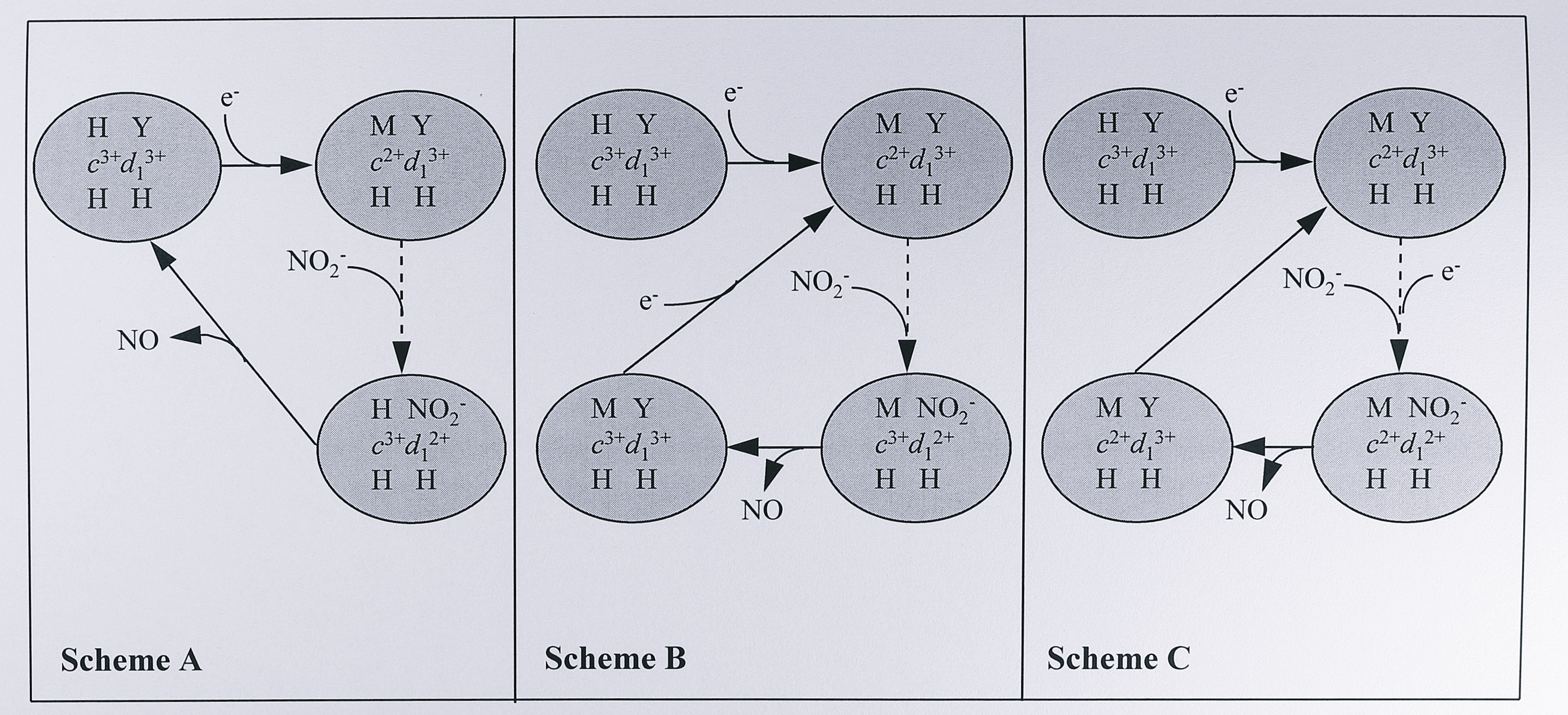
These data raise the possibility that the physiologically active form of T. pantotropha cytochrome cd\(_1\) is Met-His ligated at the c haem at all times, and that the fully-oxidised His His ligated species is a relaxed or resting artefact which is not returned to in the catalytic cycle. Three alternative reaction schemes are outlined in Figure 3.10.
In the first, the c haem returns to His-His ligation when it becomes reoxidised following intramolecular electron transfer to the d\(_1\) haem. In the second, the c haem remains Met-His ligated after reoxidation and a fully-oxidised His-His species is not regenerated. In the third, a fully oxidised enzyme is not regenerated because re-reduction of the c haem following the first intramolecular electron transfer to the d\(_1\) haem is very rapid, preventing the formation of the oxidised His-His ligation state. Further data using reduced crystals of the T. pantotropha enzyme [166] has provided some insight into which of these schemes may be correct. It has proved possible to soak the reduced crystals in potassium nitrite and then rapidly freeze them, capturing what are proposed to be structural intermediates in the catalytic cycle. The fully-reduced enzyme molecules in the crystal carry two electrons, which allows two turnovers of the enzyme. The freeze-quenched structures contain molecules captured after the binding of the second nitrite molecule and different structures show either nitrite or nitric oxide bound at the d\(_1\) haem. It is clear from the diffraction data that following the reduction of nitrite to nitric oxide at the d\(_1\) haem, the c haem has returned to His-His ligation, as in Figure 3.10a. Such a rearrangement of the haem ligands during catalysis was unexpected and it has been suggested that this is the rate-limiting step of the reaction [166]. It is also suggested that the mechanism prevents reduction of the d\(_1\) haem whilst NO is still bound; as His-17 and Tyr 25 are on the same stretch of the polypeptide chain, return of His-17 to the c haem would facilitate the re-ligation of the d\(_1\) haem by Tyr-25, displacing the bound NO. The c haem would then be re-reduced, change to Met-His ligation and transfer its electron to the d\(_1\) haem where Tyr-25 would be displaced to begin a new catalytic cycle.
However, the data from the experiments of Williams [166] are not complete. Crystalline cd\(_1\) molecules can undergo only two turnovers whereas in the periplasm it is possible that a constant supply of electrons from donor proteins would act to keep the c haem reduced and in the Met-His state. Notably, stopped flow experiments using the enzyme from Ps. aeruginosa showed that the c haem was more than 90% reduced at all times [162]. The conservation of Met-106 in all cytochrome cd\(_1\) sequences is also a strong argument for this residue to be more functionally important than His-17 in T. pantotropha cytochrome cd\(_1\). The issue will ultimately be resolved by further kinetic studies of cytochrome cd\(_1\), in conjunction with site-directed mutagenesis of His-17 and Met-106.
The role of the non-conserved Tyr-25 d\(_1\) haem ligand remains more problematic. It is clearly a ligand to the c haem in the oxidised crystal structure and can also be seen near the haem, having moved away, in the reduced state. Notably however, it has not yet been observed to return to the d\(_1\) haem in the crystal structure after NO release; this step remains hypothetical. Therefore at present it is not possible to say with certainty whether Tyr-25 plays a crucial role in the mechanism of T. pantotropha cytochrome cd\(_1\). The lack of sequence conservation in an otherwise highly homologous group of proteins does suggest that Tyr-25 may not be an obligatory residue. Alternatively we have to accept that for some reason, within a set of enzymes that are so strongly conserved as to suggest high structural homology, different functional residues are used for catalysis.
The clearest way to resolve this issue, as with the problem of haem ligation in the c domain, will be using site-directed mutagenesis to generate changes at Tyr-25 and analysis of the resultant proteins. As with all site-directed mutagenesis experiments, care will have to be taken to distinguish effects arising due to gross perturbation of the protein tertiary structure from those that involve only alteration around the haem ligation site. Bearing this in mind, obvious candidates for substitution at Tyr-25 are (1) phenylalanine, the same basic structure but lacking the liganding -OH moiety, (2) serine, of small size with an -OH group and (3) histidine, of similar size with the possibility of acting as a ligand, having been tentatively identified in other cytochromes cd\(_1\). This work is currently underway in our laboratory and some early data are presented in Chapter 6. Further crystal structures of other cytochromes cd\(_1\) will also be of great interest. The purified cytochrome cd\(_1\) from Ps. stutzeri Zobell, described in Section 3.4.3, was used for crystallisation trials by Dr. Vilmos Fülöp in the Laboratory of Molecular Biophysics and gave small crystals, though not of diffraction quality. However, we have since learned that the group of Dr. E.N. Baker (Massey University, New Zealand) is close to solving this structure, which should be available in the near future. Very recently the structure of cytochrome cd\(_1\) from Ps. aeruginosa has been determined (V. Fülöp, personal communication), although the details have not yet been published. Some aspects of this structure are discussed in Chapter 8.

![Predicted cleavage site of the periplasmic targeting sequence in cytochrome _cd_$_1$ from _T. pantotropha_. The SignalP program (Nielsen _et al_., 1997 [@nielsen_identification_1997]) predicts the cleavage site on the basis of three scores, termed C, S and Y. The residue lying immediately after the cleavage site has high C and Y scores and a sharp gradient in the S score. In the above figure it can be seen that the residue Q30 is predicted to be this residue. As described in the text, an N-terminal glutamine as the first residue of the mature protein is fully consistent with experimental data.](figure/fig3-3.png)
![Multiple alignment of cytochrome _cd_$_1$ sequences. Six previously published cytochrome _cd_$_1$ sequences (see main text for details) were downloaded from the GenBank and SwissProt databases. The sequence of cytochrome _cd_$_1$ from _T. pantotropha_ was added to the group. All sequences were edited to remove the predicted periplasmic targeting sequence and aligned using the Program ClustalW [@thompson_clustal_1994]. The multiple alignment was then processed using the program ALSCRIPT [@barton_alscript_1993], to shade homologous regions and other features of interest. Identity within four or more sequences is indicated by black shading; similarity is indicated by levels of grey shading based on the physicochemical properties of the amino acids. Conserved methionine and histidine residues which could be potential haem ligands are indicated in white on dark-grey and conserved aspartate residues at the end of the third strand in each $\beta$ propeller (see Appendix A) are shown in white on light-grey. The haem ligands His-17, His-69 (_c_-haem), His-200 and Tyr-25 (_d_$_1$ haem) in the oxidised _T. pantotropha_ enzyme are shown by open boxes. The secondary structure elements of the _T. pantotropha_ enzyme [@fulop_anatomy_1995] are shown above the alignment. Abbreviations: tp9263, _T. pantotropha_ LMD 92.63; pd1222, _P. denitrificans_ PD1222; pd12442, _P. denitrificans_ IFO 12442; pss14405, _Ps. stutzeri_ Zobell; pss300, _Ps. stutzeri_ JM300; psa6750, _Ps. aeruginosa_ NTCC 6750; aeh16, _A. eutrophus_ H16.](figure/fig3-4.png)
![Phylogenetic tree of cytochrome _cd_$_1$ sequences compared with a phylogenetic tree of 16S rRNA sequences from the same organisms. (a) The sequences were aligned using the ClustalW program and the tree was calculated using the neighbour-joining method of Saitou and Nei [@saitou_neighbor-joining_1987]. The tree was then analysed using the method of bootstrapping, with 1000 trials and visualised with the program TreeView. Sequence abbreviations are as in Table 3.3. (b) The 16S rRNA tree was calculated in the same way as for the cytochromes _cd_$_1$, except that gaps were ignored due to the partial nature of the _T. pantotropha_ LMD 92.63 and _P. denitrificans_ PD1222 sequences (obtained from Dr. D.J. Richardson, University of East Anglia). Complete 16S rRNA gene sequences for _Ps. stutzeri_ ATCC 14405, _Ps. stutzeri_ JM300, _Ps. aeruginosa_ DSM 50071 (Psa50071) and _P. denitrificans_ LMG 4218 (Pd4218) were taken from the GenBank database.](figure/fig3-5.png)
![Putative NNR binding sites in the upstream regions of the _nirS_ gene aligned with the FNR consensus binding site. The NNR boxes upstream of _nirS_ in _T. pantotropha_ LMD 92.63 (Tp9263), _P. denitrificans_ PD1222 (Pd1222) and _P. denitrificans_ IFO 12442 (Pd12442) are shown as part of a larger inverted repeat, aligned to the consensus FNR binding site derived from anaerobically-regulated _E. coli_ genes [@spiro_fnr_1994].](figure/fig3-6.png)